FlashDeconv: Fast Spatial Deconvolution via Structure-Preserving Sketching¶
This notebook demonstrates how to use FlashDeconv for spatial transcriptomics cell type deconvolution through the unified omicverse.space.Deconvolution API.
Why FlashDeconv?¶
Scalability: Handles millions of spots (Visium HD, Slide-seq) without GPU requirement
Speed: Uses randomized sketching for O(n) complexity instead of O(n²)
Spatial awareness: Incorporates graph Laplacian regularization for spatially smooth results
Integration: scanpy-style API, seamlessly works with AnnData objects
Inputs and Outputs¶
Inputs:
Spatial transcriptomics data (10x Visium, Visium HD, Slide-seq, etc.)
Single-cell reference with cell type annotations
Outputs:
Cell type proportions per spot (stored in
adata.obsm['flashdeconv'])Dominant cell type per spot
Compatible
adata_cell2locationobject for downstream analysis
Workflow Overview¶
Load scRNA-seq reference and spatial data (~1 min)
Run FlashDeconv deconvolution (~2-5 min for standard Visium)
Visualize results (~5 min)
import omicverse as ov
import scanpy as sc
import matplotlib.pyplot as plt
ov.plot_set(font_path='Arial')
🔬 Starting plot initialization...
Using already downloaded Arial font from: /tmp/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 1.7.9rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
Step 1: Load Data¶
1.1 Load scRNA-seq reference¶
The reference should contain cell type annotations in .obs.
# Load your scRNA-seq reference
# Example: Human lymph node reference
adata_sc = ov.datasets.sc_ref_Lymph_Node()
# Check cell type annotations
print(adata_sc.obs['Subset'].value_counts())
Subset
B_mem 13476
B_naive 8924
T_CD4+_naive 6012
B_Cycling 4765
T_CD4+_TfH 4690
T_CD8+_cytotoxic 3890
T_CD4+_TfH_GC 3653
B_activated 3575
B_GC_LZ 3298
T_CD4+ 3059
T_Treg 2958
B_GC_DZ 2500
T_CD8+_CD161+ 2294
T_CD8+_naive 2253
NK 1372
B_plasma 1094
T_TfR 1065
NKT 896
Endo 622
ILC 617
B_preGC 404
T_TIM3+ 357
Monocytes 306
DC_pDC 226
B_IFN 199
DC_cDC2 173
Macrophages_M1 121
Macrophages_M2 110
DC_cDC1 101
FDC 76
B_GC_prePB 74
DC_CCR7+ 42
VSMC 40
Mast 18
Name: count, dtype: int64
1.2 Load spatial transcriptomics data¶
# Load spatial data (example: Visium human lymph node)
adata_sp = sc.datasets.visium_sge(sample_id="V1_Human_Lymph_Node")
adata_sp.obs['sample'] = list(adata_sp.uns['spatial'].keys())[0]
adata_sp.var_names_make_unique()
print(f"Spatial data: {adata_sp.n_obs} spots, {adata_sp.n_vars} genes")
reading /scratch/users/steorra/analysis/omic_test/data/V1_Human_Lymph_Node/filtered_feature_bc_matrix.h5
(0:00:00)
Spatial data: 4035 spots, 36601 genes
Step 2: Run FlashDeconv Deconvolution¶
FlashDeconv is integrated into the omicverse.space.Deconvolution class. Simply set method='FlashDeconv'.
Key Parameters¶
sketch_dim: Dimension of sketched space (default: 512). Higher values preserve more information.lambda_spatial: Spatial regularization strength (default: 5000). Higher values encourage smoother spatial patterns.n_hvg: Number of highly variable genes to use (default: 2000).n_markers_per_type: Number of marker genes per cell type (default: 50).
# Initialize the Deconvolution object
decov_obj = ov.space.Deconvolution(
adata_sc=adata_sc,
adata_sp=adata_sp
)
# Run FlashDeconv deconvolution
decov_obj.deconvolution(
method='FlashDeconv',
celltype_key_sc='Subset', # Column containing cell type annotations
flashdeconv_kwargs={
'sketch_dim': 512, # Sketch dimension
'lambda_spatial': 10.0, # Spatial regularization
'n_hvg': 3000, # Number of HVGs
'n_markers_per_type': 50, # Markers per cell type
}
)
Running FlashDeconv with parameters: {'sketch_dim': 512, 'lambda_spatial': 10.0, 'n_hvg': 3000, 'n_markers_per_type': 50}
✓ FlashDeconv deconvolution is done
The deconvolution result is saved in self.adata_cell2location
Cell type proportions are also stored in self.adata_sp.obsm['flashdeconv']
Access Results¶
Results are stored in multiple locations for compatibility:
decov_obj.adata_cell2location: AnnData with cell type proportions as X matrixdecov_obj.adata_sp.obsm['flashdeconv']: DataFrame of proportionsdecov_obj.adata_sp.obs['flashdeconv_dominant']: Dominant cell type per spot
# View the result object
decov_obj.adata_cell2location
AnnData object with n_obs × n_vars = 4035 × 34
obs: 'in_tissue', 'array_row', 'array_col', 'sample', 'flashdeconv_B_Cycling', 'flashdeconv_B_GC_DZ', 'flashdeconv_B_GC_LZ', 'flashdeconv_B_GC_prePB', 'flashdeconv_B_IFN', 'flashdeconv_B_activated', 'flashdeconv_B_mem', 'flashdeconv_B_naive', 'flashdeconv_B_plasma', 'flashdeconv_B_preGC', 'flashdeconv_DC_CCR7+', 'flashdeconv_DC_cDC1', 'flashdeconv_DC_cDC2', 'flashdeconv_DC_pDC', 'flashdeconv_Endo', 'flashdeconv_FDC', 'flashdeconv_ILC', 'flashdeconv_Macrophages_M1', 'flashdeconv_Macrophages_M2', 'flashdeconv_Mast', 'flashdeconv_Monocytes', 'flashdeconv_NK', 'flashdeconv_NKT', 'flashdeconv_T_CD4+', 'flashdeconv_T_CD4+_TfH', 'flashdeconv_T_CD4+_TfH_GC', 'flashdeconv_T_CD4+_naive', 'flashdeconv_T_CD8+_CD161+', 'flashdeconv_T_CD8+_cytotoxic', 'flashdeconv_T_CD8+_naive', 'flashdeconv_T_TIM3+', 'flashdeconv_T_TfR', 'flashdeconv_T_Treg', 'flashdeconv_VSMC', 'flashdeconv_dominant'
uns: 'spatial', 'flashdeconv_params'
obsm: 'spatial', 'flashdeconv'
# View cell type proportions
decov_obj.adata_sp.obsm['flashdeconv'].head()
| B_Cycling | B_GC_DZ | B_GC_LZ | B_GC_prePB | B_IFN | B_activated | B_mem | B_naive | B_plasma | B_preGC | ... | T_CD4+_TfH | T_CD4+_TfH_GC | T_CD4+_naive | T_CD8+_CD161+ | T_CD8+_cytotoxic | T_CD8+_naive | T_TIM3+ | T_TfR | T_Treg | VSMC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAACAAGTATCTCCCA-1 | 0.000000 | 0.142163 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.085327 | 0.390464 | 0.0 | ... | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.00000 | 0.0 | 0.040701 |
| AAACAATCTACTAGCA-1 | 0.000000 | 0.163051 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.116814 | 0.0 | ... | 0.0 | 0.049438 | 0.176642 | 0.0 | 0.0 | 0.005135 | 0.000000 | 0.15238 | 0.0 | 0.026961 |
| AAACACCAATAACTGC-1 | 0.016557 | 0.130161 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.046586 | 0.232831 | 0.0 | ... | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.013086 | 0.00000 | 0.0 | 0.054721 |
| AAACAGAGCGACTCCT-1 | 0.000000 | 0.172508 | 0.0 | 0.0 | 0.225865 | 0.0 | 0.0 | 0.000000 | 0.078707 | 0.0 | ... | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.126967 | 0.00000 | 0.0 | 0.000000 |
| AAACAGCTTTCAGAAG-1 | 0.000000 | 0.150919 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.403057 | 0.133602 | 0.0 | ... | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.00000 | 0.0 | 0.000000 |
5 rows × 34 columns
Step 3: Visualization¶
3.1 Spatial heatmap of cell type proportions¶
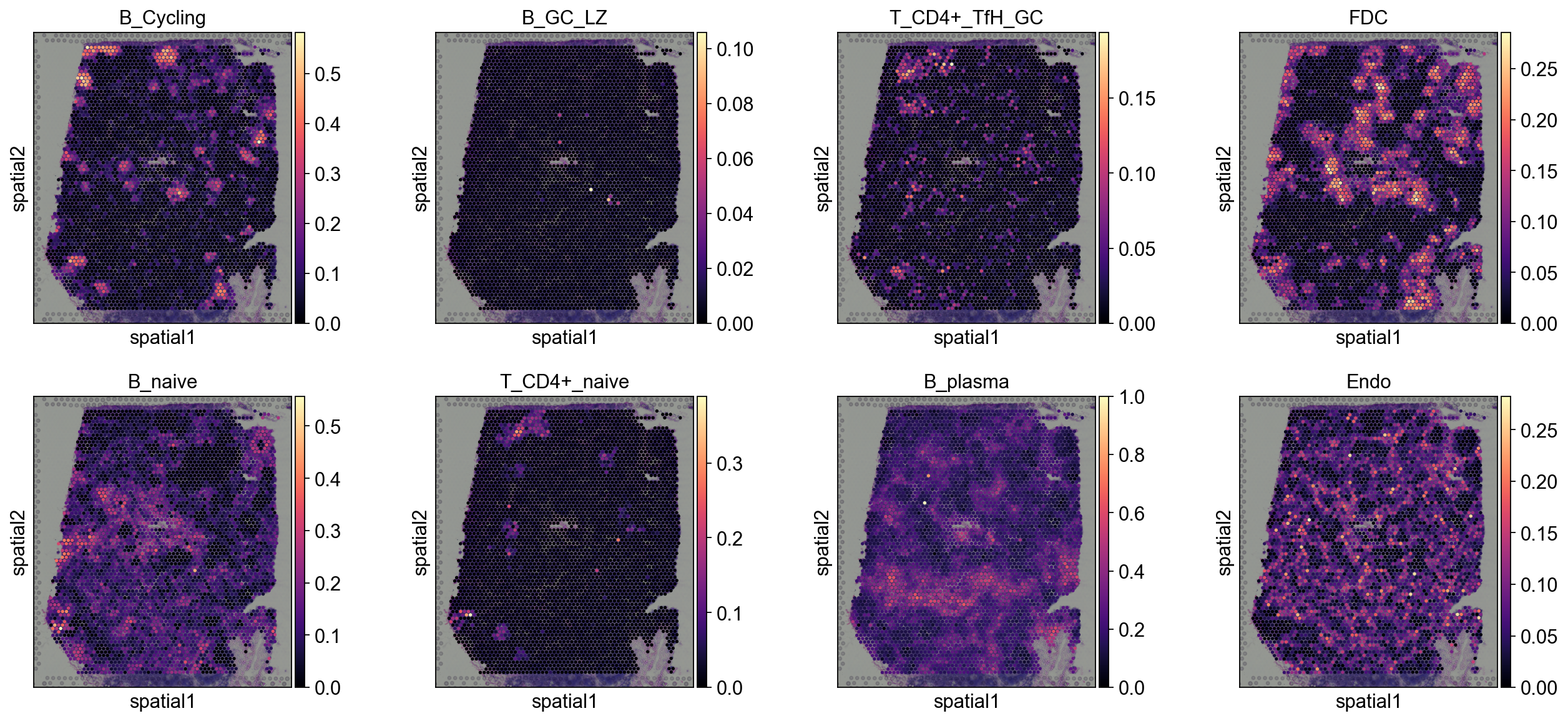
# Select cell types to visualize
annotation_list=['B_Cycling', 'B_GC_LZ', 'T_CD4+_TfH_GC', 'FDC',
'B_naive', 'T_CD4+_naive', 'B_plasma', 'Endo']
# Plot spatial distribution
sc.pl.spatial(
decov_obj.adata_cell2location,
cmap='magma',
color=annotation_list,
ncols=4,
size=1.3,
img_key='hires',
)
3.2 Dominant cell type visualization¶
# Plot dominant cell type per spot
sc.pl.spatial(
decov_obj.adata_sp,
color='flashdeconv_dominant',
size=1.3,
img_key='hires',
)

3.3 Multi-target overlay¶
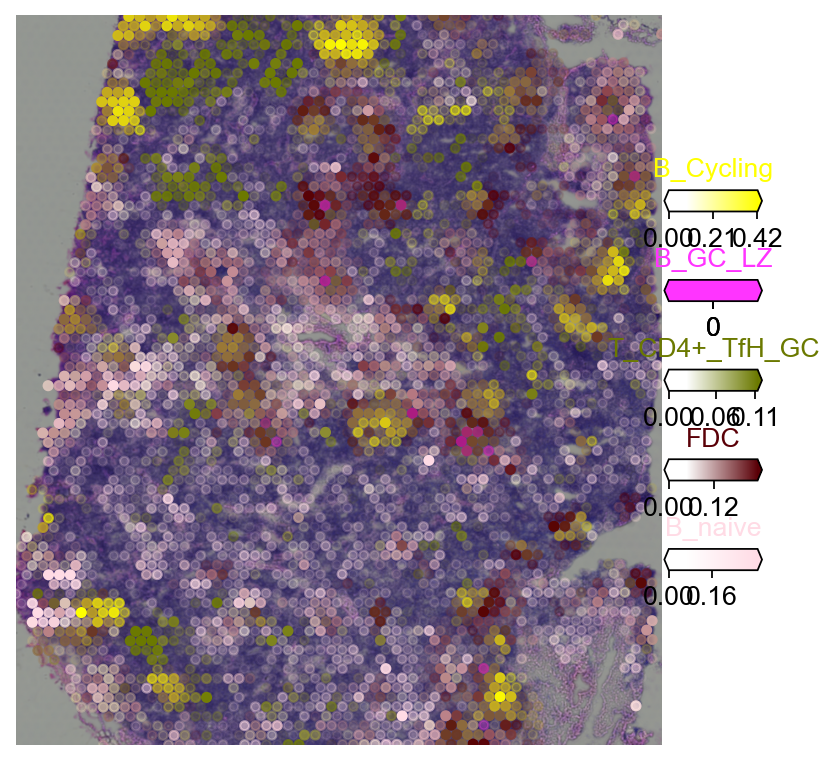
import matplotlib as mpl
# Create color dictionary from reference
if 'Subset_colors' in adata_sc.uns:
color_dict = dict(zip(
adata_sc.obs['Subset'].cat.categories,
adata_sc.uns['Subset_colors']
))
else:
color_dict = None
clust_labels = annotation_list[:5]
with mpl.rc_context({'figure.figsize': (6, 6), 'axes.grid': False}):
fig = ov.pl.plot_spatial(
adata=decov_obj.adata_cell2location,
color=clust_labels,
labels=clust_labels,
show_img=True,
style='fast',
max_color_quantile=0.992,
circle_diameter=4,
colorbar_position='right',
palette=color_dict
)
3.4 Pie chart visualization (cropped region)¶
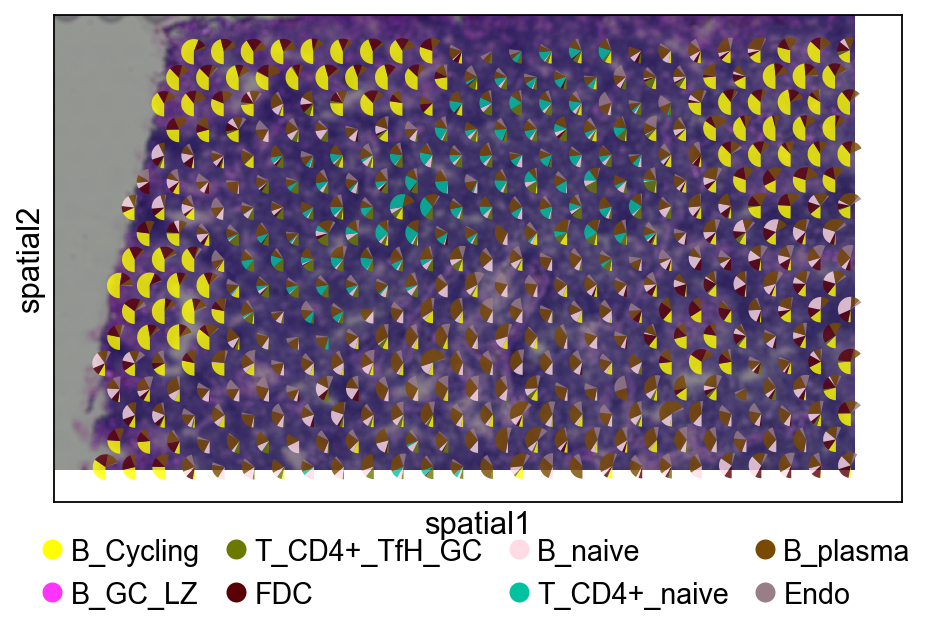
# Crop a region of interest
adata_cropped = ov.space.crop_space_visium(
decov_obj.adata_cell2location,
crop_loc=(0, 0),
crop_area=(500, 1000),
library_id=list(decov_obj.adata_cell2location.uns['spatial'].keys())[0],
scale=1
)
# Plot with pie charts
fig, ax = plt.subplots(figsize=(8, 4))
sc.pl.spatial(
adata_cropped,
basis='spatial',
color=None,
size=1.3,
img_key='hires',
ax=ax,
show=False
)
ov.pl.add_pie2spatial(
adata_cropped,
img_key='hires',
cell_type_columns=annotation_list,
ax=ax,
colors=color_dict,
pie_radius=10,
remainder='gap',
legend_loc=(0.5, -0.25),
ncols=4,
alpha=0.8
)
plt.show()
Adding image layer `image`
Comparison: FlashDeconv vs Other Methods¶
Feature |
FlashDeconv |
Tangram |
cell2location |
|---|---|---|---|
GPU Required |
No |
Optional |
Recommended |
Speed (10k spots) |
~2 min |
~15 min |
~60 min |
Visium HD Support |
Yes (native) |
Limited |
Limited |
Spatial Regularization |
Built-in |
No |
No |
API Style |
scanpy-like |
Custom |
Custom |
Tips and Troubleshooting¶
Parameter Tuning¶
For noisy/sparse data: Increase
lambda_spatial(e.g., 10000)For dense data (Visium HD 2μm): Increase
sketch_dim(e.g., 1024)For better accuracy: Increase
n_hvg(e.g., 3000)
Common Issues¶
Few overlapping genes: Ensure gene names match between spatial and reference data
Missing spatial coordinates: Check
adata.obsm['spatial']existsMemory issues with large data: FlashDeconv is memory-efficient, but for very large datasets, consider subsetting
Citation¶
If you use FlashDeconv in your research, please cite:
Yang, C., Chen, J. & Zhang, X. FlashDeconv enables atlas-scale,
multi-resolution spatial deconvolution via structure-preserving sketching.
bioRxiv (2025). https://doi.org/10.64898/2025.12.22.696108
Also cite OmicVerse for the unified API:
Zeng, Z., et al. OmicVerse: a framework for bridging and accelerating
single-cell multiomics analysis with deep learning.