Back to top
Preprocessing the data of scRNA-seq with omicverse[CPU-GPU-mixed]
The count table, a numeric matrix of genes × cells, is the basic input data structure in the analysis of single-cell RNA-sequencing data. A common preprocessing step is to adjust the counts for variable sampling efficiency and to transform them so that the variance is similar across the dynamic range.
Suitable methods to preprocess the scRNA-seq is important. Here, we introduce some preprocessing step to help researchers can perform downstream analysis easyier.
User can compare our tutorial with scanpy’tutorial to learn how to use omicverse well
Colab_Reproducibility:https://colab.research.google.com/drive/1DXLSls_ppgJmAaZTUvqazNC_E7EDCxUe?usp=sharing
Note
“When OmicVerse is upgraded to version > 1.7.0, it supports CPU–GPU mixed acceleration without requiring rapids_singlecell
The data consist of 3k PBMCs from a Healthy Donor and are freely available from 10x Genomics (here from this webpage ). On a unix system, you can uncomment and run the following to download and unpack the data. The last line creates a directory for writing processed data.
Preprocessing
Quantity control
For single-cell data, we require quality control prior to analysis, including the removal of cells containing double cells, low-expressing cells, and low-expressing genes. In addition to this, we need to filter based on mitochondrial gene ratios, number of transcripts, number of genes expressed per cell, cellular Complexity, etc. For a detailed description of the different QCs please see the document: https://hbctraining.github.io/scRNA-seq/lessons/04_SC_quality_control.html
Note
if the version of omicverse 1.6.4 doublets_method scrublet sccomposite
COMPOSITE (COMpound POiSson multIplet deTEction model) is a computational tool for multiplet detection in both single-cell single-omics and multiomics settings. It has been implemented as an automated pipeline and is available as both a cloud-based application with a user-friendly interface and a Python package.
Hu, H., Wang, X., Feng, S. et al. A unified model-based framework for doublet or multiplet detection in single-cell multiomics data. Nat Commun 15, 5562 (2024). https://doi.org/10.1038/s41467-024-49448-x
⚙️ Using CPU/GPU mixed mode for QC...
NVIDIA CUDA GPUs detected:
📊 [CUDA 0] NVIDIA L40S
------------------------------ 4/46068 MiB (0.0%)
🔍 Quality Control Analysis (CPU-GPU Mixed):
Dataset shape: 2,700 cells × 32,738 genes
QC mode: seurat
Doublet detection: scrublet
Mitochondrial genes: MT-
📊 Step 1: Calculating QC Metrics
✓ Gene Family Detection:
┌──────────────────────────────┬────────────────────┬────────────────────┐
│ Gene Family │ Genes Found │ Detection Method │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Mitochondrial │ 13 │ Auto (MT-) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Ribosomal │ 106 │ Auto (RPS/RPL) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Hemoglobin │ 13 │ Auto (regex) │
└──────────────────────────────┴────────────────────┴────────────────────┘
✓ QC Metrics Summary:
┌─────────────────────────┬────────────────────┬─────────────────────────┐
│ Metric │ Mean │ Range (Min - Max) │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ nUMIs │ 2367 │ 548 - 15844 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Detected Genes │ 847 │ 212 - 3422 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Mitochondrial % │ 2.2% │ 0.0% - 22.6% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Ribosomal % │ 34.9% │ 1.1% - 59.4% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Hemoglobin % │ 0.0% │ 0.0% - 1.4% │
└─────────────────────────┴────────────────────┴─────────────────────────┘
🔧 Step 2: Quality Filtering (SEURAT)
Thresholds: mito≤0.2, nUMIs≥500, genes≥250
📊 Seurat Filter Results:
• nUMIs filter (≥500): 0 cells failed (0.0%)
• Genes filter (≥250): 3 cells failed (0.1%)
• Mitochondrial filter (≤0.2): 2 cells failed (0.1%)
✓ Combined QC filters: 2,695 cells removed (99.8%)
🎯 Step 3: Final Filtering
Parameters: min_genes=200, min_cells=3
Ratios: max_genes_ratio=1, max_cells_ratio=1
✓ Final filtering: 0 cells, 19,024 genes removed
🔍 Step 4: Doublet Detection
⚠️ Note: 'scrublet' detection is legacy and may not work optimally
💡 Consider using 'doublets_method=sccomposite' for better results
🔍 Running scrublet doublet detection...
🔍 Running Scrublet Doublet Detection:
Mode: cpu-gpu-mixed
Computing doublet prediction using Scrublet algorithm
🔍 Filtering genes and cells...
🔍 Filtering genes...
Parameters: min_cells≥3
✓ Filtered: 0 genes removed
🔍 Filtering cells...
Parameters: min_genes≥3
✓ Filtered: 0 cells removed
🔍 Normalizing data and selecting highly variable genes...
🔍 Count Normalization:
Target sum: median
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 2,695 cells × 13,714 genes
✓ Runtime: 0.01s
🔍 Highly Variable Genes Selection:
Method: seurat
⚠️ Gene indices [7854] fell into a single bin: normalized dispersion set to 1
💡 Consider decreasing `n_bins` to avoid this effect
✅ HVG Selection Completed Successfully!
✓ Selected: 1,739 highly variable genes out of 13,714 total (12.7%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'dispersions': Float vector (adata.var)
• 'dispersions_norm': Float vector (adata.var)
🔍 Simulating synthetic doublets...
🔍 Normalizing observed and simulated data...
🔍 Count Normalization:
Target sum: 1000000.0
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 2,695 cells × 1,739 genes
✓ Runtime: 0.00s
🔍 Count Normalization:
Target sum: 1000000.0
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 5,390 cells × 1,739 genes
✓ Runtime: 0.01s
🔍 Embedding transcriptomes using PCA...
Using CUDA device: NVIDIA L40S
✅ Using built-in torch_pca for GPU-accelerated PCA
🚀 Using torch_pca PCA for CUDA GPU acceleration in scrublet
🚀 torch_pca PCA backend: CUDA GPU acceleration (scrublet, supports sparse)
📊 Scrublet PCA input data type - X_obs: ndarray, shape: (2695, 1739), dtype: float64
📊 Scrublet PCA input data type - X_sim: ndarray, shape: (5390, 1739), dtype: float64
🔍 Calculating doublet scores...
🔍 Calling doublets with threshold detection...
📊 Automatic threshold: 0.309
📈 Detected doublet rate: 1.3%
🔍 Detectable doublet fraction: 33.5%
📊 Overall doublet rate comparison:
• Expected: 5.0%
• Estimated: 3.8%
✅ Scrublet Analysis Completed Successfully!
✓ Results added to AnnData object:
• 'doublet_score': Doublet scores (adata.obs)
• 'predicted_doublet': Boolean predictions (adata.obs)
• 'scrublet': Parameters and metadata (adata.uns)
✓ Scrublet completed: 34 doublets removed (1.3%)
╭─ SUMMARY: qc ──────────────────────────────────────────────────────╮
│ Duration: 10.991s │
│ Shape: 2,700 x 32,738 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBS │ ✚ cell_complexity (float) │
│ │ ✚ detected_genes (int) │
│ │ ✚ hb_perc (float) │
│ │ ✚ mito_perc (float) │
│ │ ✚ nUMIs (float) │
│ │ ✚ passing_mt (bool) │
│ │ ✚ passing_nUMIs (bool) │
│ │ ✚ passing_ngenes (bool) │
│ │ ✚ passing_qc_step1 (bool) │
│ │ ✚ ribo_perc (float) │
│ │
│ ● VAR │ ✚ hb (bool) │
│ │ ✚ mt (bool) │
│ │ ✚ ribo (bool) │
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 16.2 s, sys: 2.92 s, total: 19.1 s
Wall time: 42.5 s
High variable Gene Detection
Here we try to use Pearson’s method to calculate highly variable genes. This is the method that is proposed to be superior to ordinary normalisation. See Article in Nature Method for details.
normalize|HVGs:We use | to control the preprocessing step, | before for the normalisation step, either shiftlog pearson pearson seurat shiftlog|pearson
if you use mode shiftlog|pearson target_sum=50*1e4 target_sum=1e4
if you use mode pearson|pearson target_sum
Note
if the version of omicverse 1.4.13 scanpy pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 0 highly-expressed genes from normalization computation
Excluded genes: []
✅ Count Normalization Completed Successfully!
✓ Processed: 2,661 cells × 13,714 genes
✓ Runtime: 0.12s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 2,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 2,000 highly variable genes out of 13,714 total (14.6%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 1.30 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 1.3415s │
│ Shape: 2,661 x 13,714 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● VAR │ ✚ highly_variable (bool) │
│ │ ✚ highly_variable_features (bool) │
│ │ ✚ highly_variable_rank (float) │
│ │ ✚ means (float) │
│ │ ✚ n_cells (int) │
│ │ ✚ percent_cells (float) │
│ │ ✚ residual_variances (float) │
│ │ ✚ robust (bool) │
│ │ ✚ variances (float) │
│ │
│ ● UNS │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │
│ ● LAYERS │ ✚ counts (sparse matrix, 2661x13714) │
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 1.45 s, sys: 126 ms, total: 1.57 s
Wall time: 1.34 s
You can use recover_counts
AAACATACAACCAC-1
AAACATTGAGCTAC-1
AAACATTGATCAGC-1
AAACCGTGCTTCCG-1
AAACCGTGTATGCG-1
AAACGCACTGGTAC-1
AAACGCTGACCAGT-1
AAACGCTGGTTCTT-1
AAACGCTGTAGCCA-1
AAACGCTGTTTCTG-1
...
TTTCAGTGTCACGA-1
TTTCAGTGTCTATC-1
TTTCAGTGTGCAGT-1
TTTCCAGAGGTGAG-1
TTTCGAACACCTGA-1
TTTCGAACTCTCAT-1
TTTCTACTGAGGCA-1
TTTCTACTTCCTCG-1
TTTGCATGAGAGGC-1
TTTGCATGCCTCAC-1
CD3D
6.718757
0.0
7.371373
0.0
0.0
5.447429
6.132899
6.499361
5.974209
0.0
...
0.0
0.0
0.0
6.532146
0.0
0.0
0.0
0.0
0.0
6.224622
1 rows × 2661 columns
AAACATACAACCAC-1
AAACATTGAGCTAC-1
AAACATTGATCAGC-1
AAACCGTGCTTCCG-1
AAACCGTGTATGCG-1
AAACGCACTGGTAC-1
AAACGCTGACCAGT-1
AAACGCTGGTTCTT-1
AAACGCTGTAGCCA-1
AAACGCTGTTTCTG-1
...
TTTCAGTGTCACGA-1
TTTCAGTGTCTATC-1
TTTCAGTGTGCAGT-1
TTTCCAGAGGTGAG-1
TTTCGAACACCTGA-1
TTTCGAACTCTCAT-1
TTTCTACTGAGGCA-1
TTTCTACTTCCTCG-1
TTTGCATGAGAGGC-1
TTTGCATGCCTCAC-1
CD3D
4.0
0.0
10.0
0.0
0.0
1.0
2.0
3.0
1.0
0.0
...
0.0
0.0
0.0
3.0
0.0
0.0
0.0
0.0
0.0
2.0
1 rows × 2667 columns
AAACATACAACCAC-1
AAACATTGAGCTAC-1
AAACATTGATCAGC-1
AAACCGTGCTTCCG-1
AAACCGTGTATGCG-1
AAACGCACTGGTAC-1
AAACGCTGACCAGT-1
AAACGCTGGTTCTT-1
AAACGCTGTAGCCA-1
AAACGCTGTTTCTG-1
...
TTTCAGTGTCACGA-1
TTTCAGTGTCTATC-1
TTTCAGTGTGCAGT-1
TTTCCAGAGGTGAG-1
TTTCGAACACCTGA-1
TTTCGAACTCTCAT-1
TTTCTACTGAGGCA-1
TTTCTACTTCCTCG-1
TTTGCATGAGAGGC-1
TTTGCATGCCTCAC-1
CD3D
4
0
9
0
0
1
2
2
1
0
...
0
0
0
2
0
0
0
0
0
1
1 rows × 2654 columns
Set the .raw attribute of the AnnData object to the normalized and logarithmized raw gene expression for later use in differential testing and visualizations of gene expression. This simply freezes the state of the AnnData object.
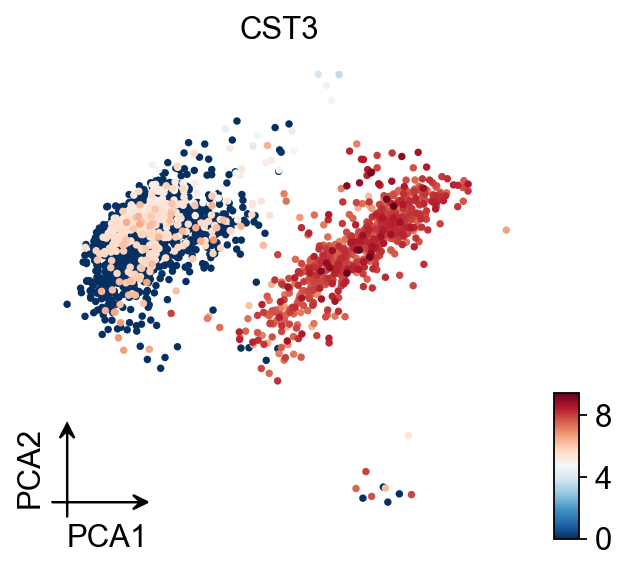
Principal component analysis
In contrast to scanpy, we do not directly scale the variance of the original expression matrix, but store the results of the variance scaling in the layer, due to the fact that scale may cause changes in the data distribution, and we have not found scale to be meaningful in any scenario other than a principal component analysis
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 0.7342s │
│ Shape: 2,661 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● LAYERS │ ✚ scaled (sparse matrix, 2661x2000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 571 ms, sys: 262 ms, total: 833 ms
Wall time: 737 ms
If you want to perform pca in normlog layer, you can set layer normlog
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 12.5752s │
│ Shape: 2,661 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ pca │
│ │ └─ params: {'zero_center': True, 'use_highly_variable': Tr... │
│ │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ X_pca (array, 2661x50) │
│ │ ✚ scaled|original|X_pca (array, 2661x50) │
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 34.1 s, sys: 1min 19s, total: 1min 53s
Wall time: 12.6 s
Embedding the neighborhood graph
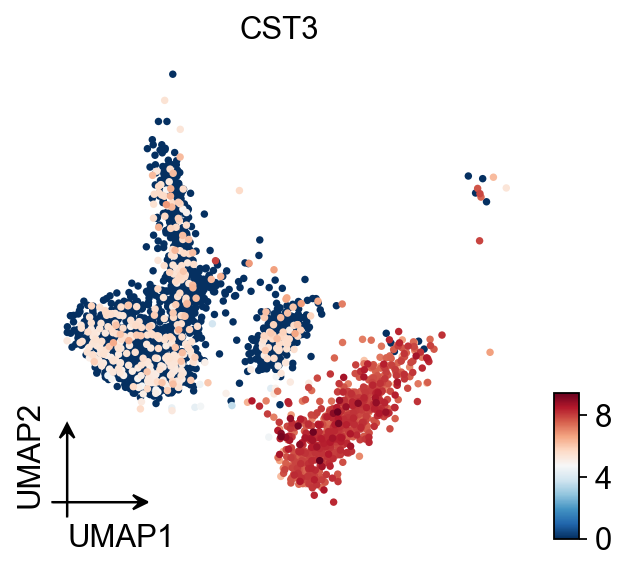
We suggest embedding the graph in two dimensions using UMAP (McInnes et al., 2018), see below. It is potentially more faithful to the global connectivity of the manifold than tSNE, i.e., it better preserves trajectories. In some ocassions, you might still observe disconnected clusters and similar connectivity violations. They can usually be remedied by running:
🔍 K-Nearest Neighbors Graph Construction:
Mode: cpu-gpu-mixed
Neighbors: 15
Method: torch
Metric: euclidean
Representation: scaled|original|X_pca
PCs used: 50
🔍 Computing neighbor distances...
🔍 Computing connectivity matrix...
💡 Using UMAP-style connectivity
✓ Graph is fully connected
✅ KNN Graph Construction Completed Successfully!
✓ Processed: 2,661 cells with 15 neighbors each
✓ Results added to AnnData object:
• 'neighbors': Neighbors metadata (adata.uns)
• 'distances': Distance matrix (adata.obsp)
• 'connectivities': Connectivity matrix (adata.obsp)
╭─ SUMMARY: neighbors ───────────────────────────────────────────────╮
│ Duration: 2.8979s │
│ Shape: 2,661 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ neighbors │
│ │ └─ params: {'n_neighbors': 15, 'method': 'torch', 'random_... │
│ │
│ ● OBSP │ ✚ connectivities (sparse matrix, 2661x2661) │
│ │ ✚ distances (sparse matrix, 2661x2661) │
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 5.8 s, sys: 150 ms, total: 5.95 s
Wall time: 2.9 s
UMAP
You also can use umap
🔍 UMAP Dimensionality Reduction:
Mode: cpu-gpu-mixed
Method: pumap
Components: 2
Min distance: 0.5
{'n_neighbors': 15, 'method': 'torch', 'random_state': 0, 'metric': 'euclidean', 'use_rep': 'scaled|original|X_pca', 'n_pcs': 50}
⚠️ Connectivities matrix was not computed with UMAP method
🔍 Computing UMAP parameters...
🔍 Computing UMAP embedding (Parametric PyTorch method)...
Using device: cuda
Dataset: 2661 samples × 50 features
Batch size: 266
Learning rate: 0.001
Training parametric UMAP model...
============================================================
🚀 Parametric UMAP Training
============================================================
📊 Device: cuda
📈 Data shape: torch.Size([2661, 50])
🔗 Building UMAP graph...
🚀 Using PyTorch Geometric KNN (faster)
🏋️ Starting Training...
────────────────────────────────────────────────────────────
✓ New best loss: 0.5572
✓ New best loss: 0.3124
✓ New best loss: 0.2870
✓ New best loss: 0.2688
✓ New best loss: 0.2632
────────────────────────────────────────────────────────────
✅ Training Completed!
📉 Final best loss: 0.2632
============================================================
💡 Using Parametric UMAP (PyTorch) on cuda
✅ UMAP Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 2,661 cells × 2 dimensions
✓ Results added to AnnData object:
• 'X_umap': UMAP coordinates (adata.obsm)
• 'umap': UMAP parameters (adata.uns)
✅ UMAP completed successfully.
╭─ SUMMARY: umap ────────────────────────────────────────────────────╮
│ Duration: 4.9374s │
│ Shape: 2,661 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ umap │
│ │ └─ params: {'a': 0.583030020479462, 'b': 1.3341669929263487} │
│ │
│ ● OBSM │ ✚ X_umap (array, 2661x2) │
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 3.88 s, sys: 430 ms, total: 4.3 s
Wall time: 4.94 s
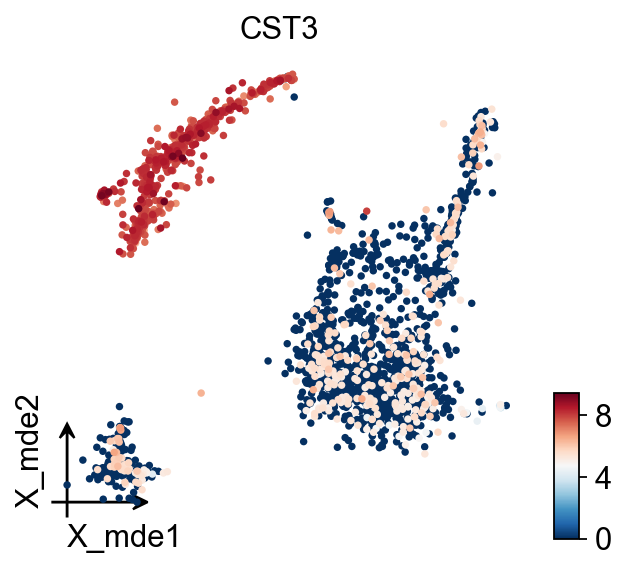
MDE
To visualize the PCA’s embeddings, we use the pymde
🔍 MDE Dimensionality Reduction:
Mode: cpu-gpu-mixed
Embedding dimensions: 2
Neighbors: 15
Repulsive fraction: 0.7
Using representation: scaled|original|X_pca
Principal components: 50
Using CUDA device: NVIDIA L40S
✅ Using built-in torch_pca for GPU-accelerated PCA
🔍 Computing k-nearest neighbors graph...
🔍 Creating MDE embedding...
🔍 Optimizing embedding...
✅ MDE Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 2,661 cells × 2 dimensions
✓ Runtime: 3.44s
✓ Results added to AnnData object:
• 'X_mde': MDE coordinates (adata.obsm)
• 'neighbors': Neighbors metadata (adata.uns)
• 'distances': Distance matrix (adata.obsp)
• 'connectivities': Connectivity matrix (adata.obsp)
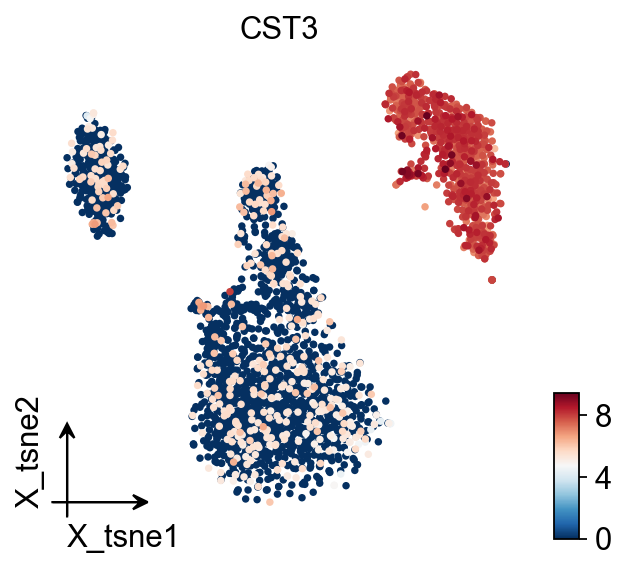
TSNE
🔍 t-SNE Dimensionality Reduction:
Mode: cpu-gpu-mixed
Components: 2
Perplexity: 30
Learning rate: 1000
🔍 Computing t-SNE with scikit-learn...
💡 Using scikit-learn TSNE implementation
✅ t-SNE Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 2,661 cells × 2 dimensions
✓ Results added to AnnData object:
• 'X_tsne': t-SNE coordinates (adata.obsm)
• 'tsne': t-SNE parameters (adata.uns)
╭─ SUMMARY: tsne ────────────────────────────────────────────────────╮
│ Duration: 31.0173s │
│ Shape: 2,661 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ tsne │
│ │ └─ params: {'perplexity': 30, 'early_exaggeration': 12, 'l... │
│ │
│ ● OBSM │ ✚ X_tsne (array, 2661x2) │
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 5min 25s, sys: 1.08 s, total: 5min 27s
Wall time: 31 s
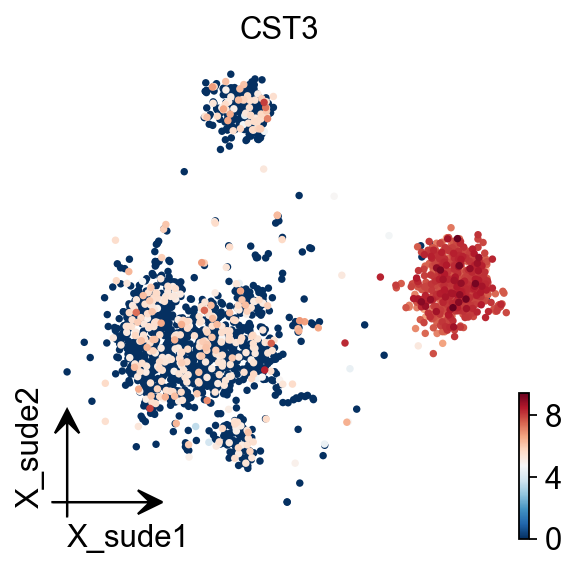
SUDE
SUDE, a scalable manifold learning method that uses uniform landmark sampling and constrained embedding to preserve both global structure and cluster separability, outperforming t-SNE, UMAP, and related methods across synthetic, real-world, and biomedical datasets.
Cite: Peng, D., Gui, Z., Wei, W. et al. Sampling-enabled scalable manifold learning unveils the discriminative cluster structure of high-dimensional data. Nat Mach Intell (2025). https://doi.org/10.1038/s42256-025-01112-9
🔍 SUDE Dimensionality Reduction:
Mode: cpu-gpu-mixed
Components: 2
Landmarks (k1): 20
Initialization: le
🔍 Computing SUDE embedding...
Parameters:
• Dimensions: 2
• K1 landmarks: 20
• Normalize: True
• Large scale: False
• Aggregation coef: 1.2
• Max epochs: 50
✅ SUDE Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 2,668 cells × 2 dimensions
✓ Results added to AnnData object:
• 'X_sude': SUDE coordinates (adata.obsm)
• 'sude': SUDE parameters (adata.uns)
CPU times: user 14.1 s, sys: 4.41 s, total: 18.5 s
Wall time: 4.42 s
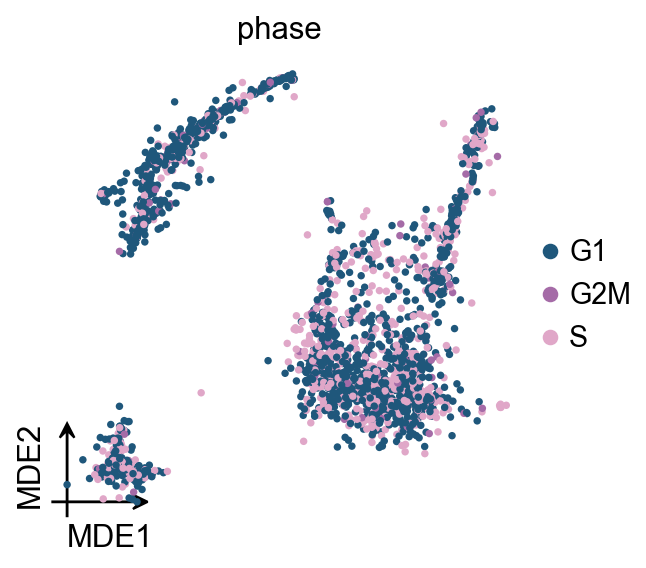
Score cell cyle
In OmicVerse, we store both G1M/S and G2M genes into the function (both human and mouse), so you can run cell cycle analysis without having to manually enter cycle genes!
╭─ SUMMARY: score_genes_cell_cycle ──────────────────────────────────╮
│ Duration: 0.1036s │
│ Shape: 2,661 x 13,714 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBS │ ✚ G2M_score (float) │
│ │ ✚ S_score (float) │
│ │ ✚ phase (str) │
│ │
╰────────────────────────────────────────────────────────────────────╯
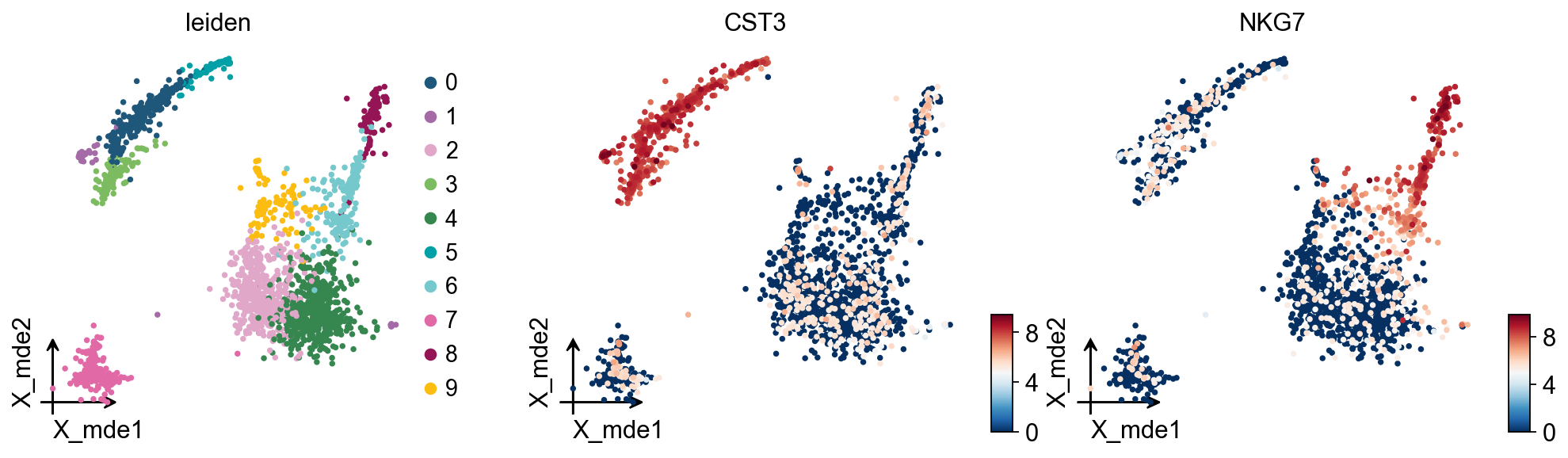
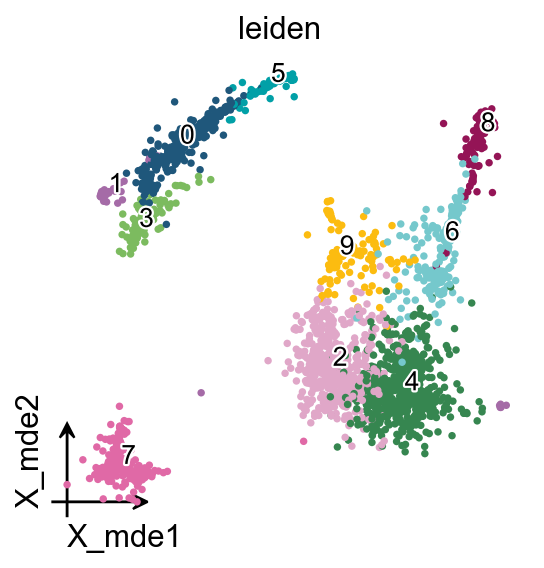
Clustering the neighborhood graph
As with Seurat and many other frameworks, we recommend the Leiden graph-clustering method (community detection based on optimizing modularity) by Traag et al. (2018). Note that Leiden clustering directly clusters the neighborhood graph of cells, which we already computed in the previous section.
╭─ SUMMARY: leiden ──────────────────────────────────────────────────╮
│ Duration: 10.078s │
│ Shape: 2,661 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBS │ ✚ leiden (category) │
│ │
│ ● UNS │ ✚ leiden │
│ │ └─ params: {'resolution': 1, 'random_state': 0, 'local_ite... │
│ │
╰────────────────────────────────────────────────────────────────────╯
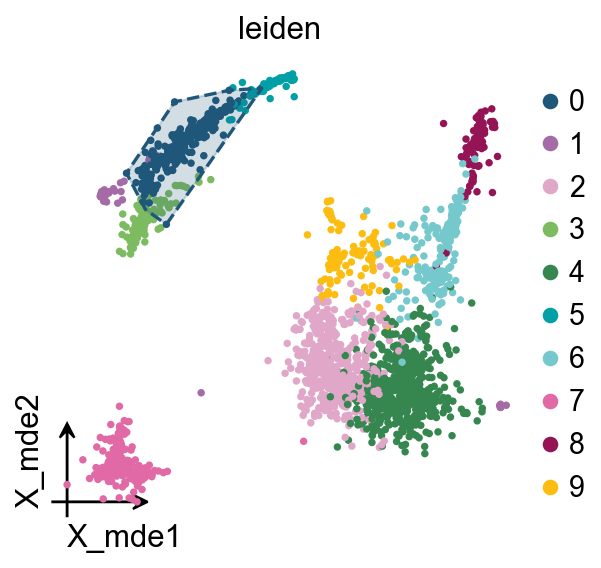
We redesigned the visualisation of embedding to distinguish it from scanpy’s embedding by adding the parameter fraemon='small'
We also provide a boundary visualisation function ov.utils.plot_ConvexHull
Arguments:
If you have too many labels, e.g. too many cell types, and you are concerned about cell overlap, then consider trying the ov.utils.gen_mpl_labels patheffects
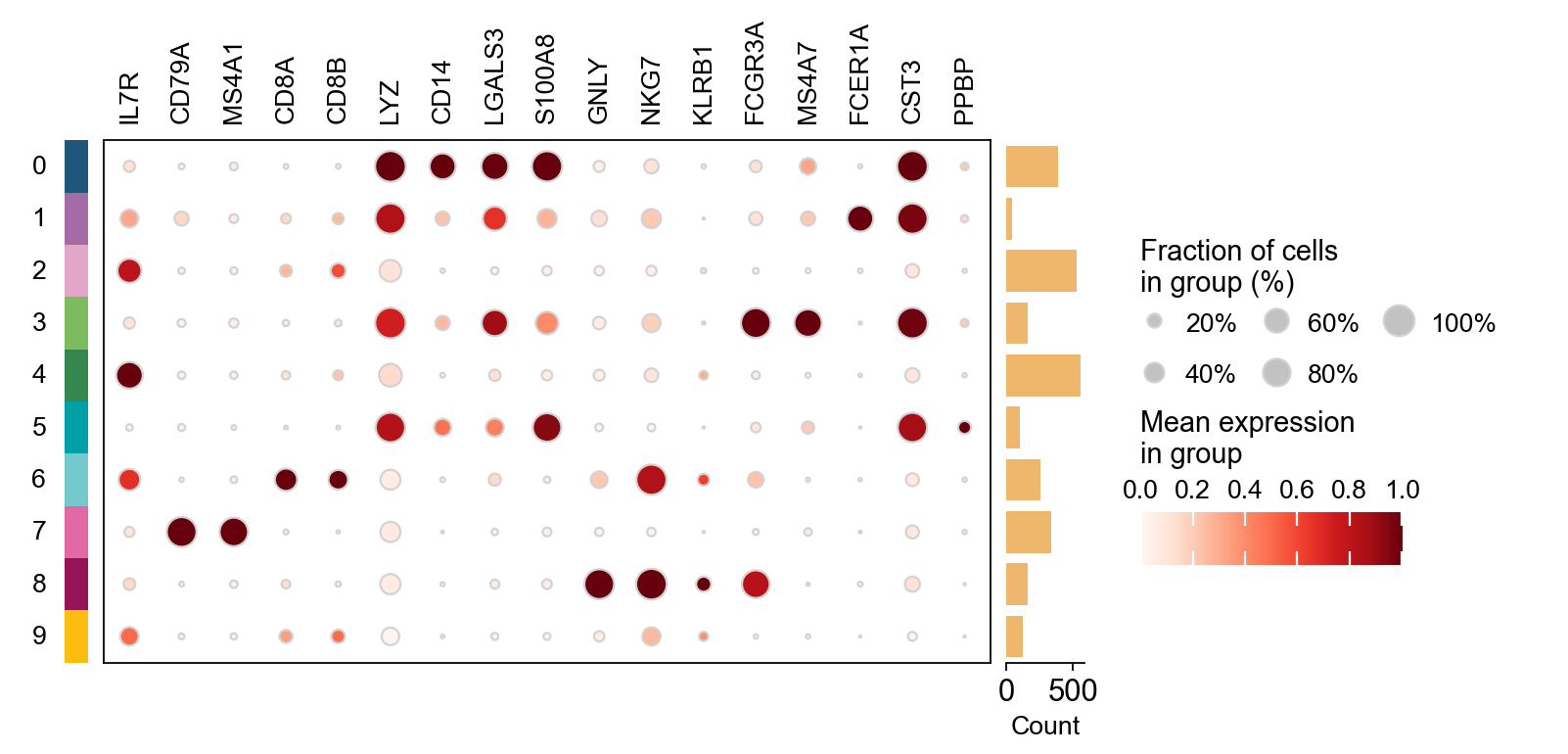
Finding marker genes
Let us compute a ranking for the highly differential genes in each cluster. For this, by default, the .raw attribute of AnnData is used in case it has been initialized before. The simplest and fastest method to do so is the t-test.
method: wilcoxon | groupby: leiden | n_groups: 10 | n_genes: 50
✅ Done | 10 groups × 50 genes | corr: benjamini-hochberg | stored in adata.uns['rank_genes_groups']
CPU times: user 2.22 s, sys: 399 ms, total: 2.62 s
Wall time: 2.62 s
group
rank
names
scores
logfoldchanges
pvals
pvals_adj
pct_group
pct_rest
0
0
1
LYZ
30.139215
8.308955
1.485183e-199
2.036780e-195
1.000000
0.536060
1
0
2
S100A9
29.859339
10.341062
6.640705e-196
4.553531e-192
0.997416
0.245822
2
0
3
S100A8
28.556858
10.218678
2.308892e-179
1.055472e-175
0.968992
0.156552
3
0
4
FCN1
28.287178
8.907052
4.969369e-176
1.703748e-172
0.976744
0.176781
4
0
5
TYROBP
28.184223
8.633690
9.127943e-175
2.503612e-171
1.000000
0.292436
5
0
6
CST3
28.036358
8.869659
5.858849e-173
1.339138e-169
1.000000
0.293316
6
0
7
LGALS2
27.883018
9.479619
4.287074e-171
8.398990e-168
0.937984
0.088830
7
0
8
S100A6
27.122936
4.239765
5.282608e-162
8.049520e-159
1.000000
0.743184
8
0
9
GSTP1
26.817543
6.501383
2.017550e-158
2.766869e-155
0.976744
0.367194
9
0
10
FTL
26.692188
3.368342
5.800019e-157
7.231042e-154
1.000000
0.985488
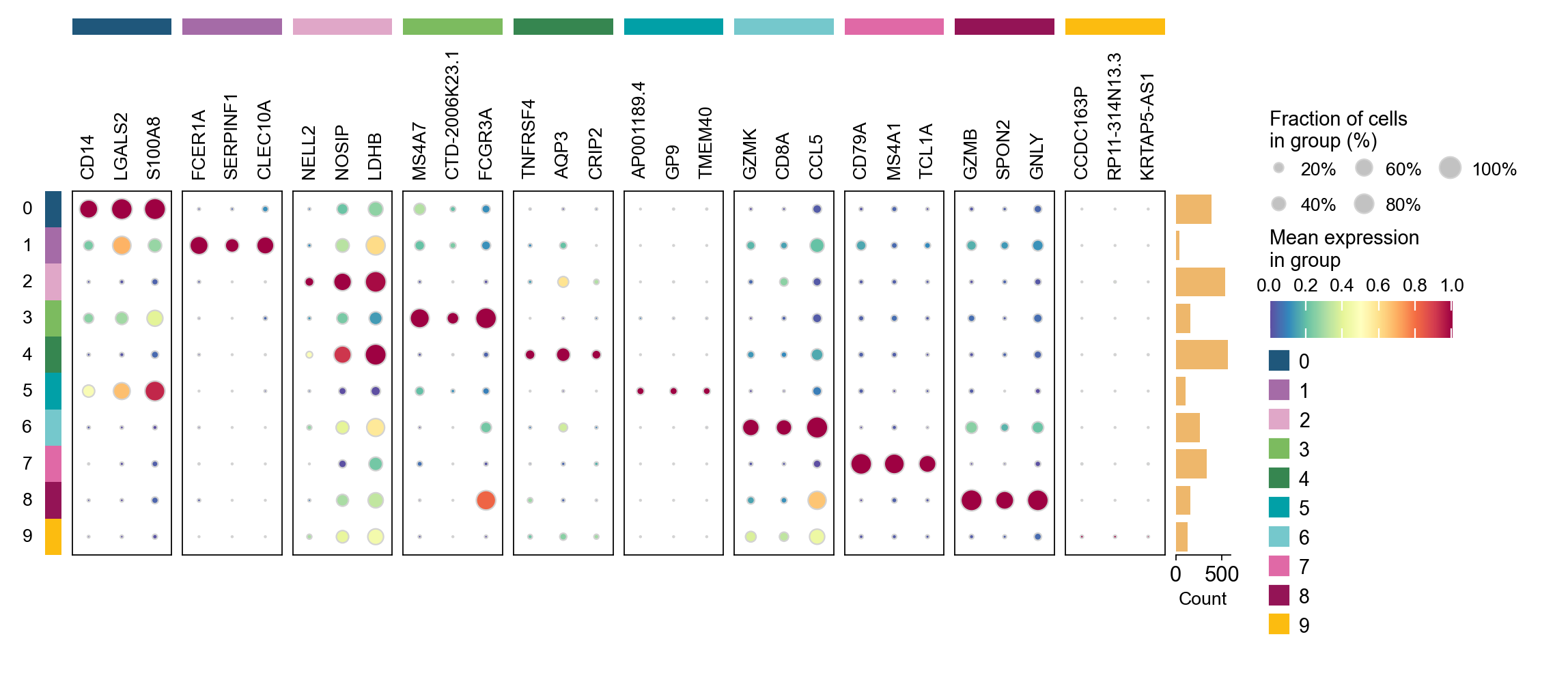
cosg is also considered to be a better algorithm for finding marker genes. Here, omicverse provides the calculation of cosg
Paper: Accurate and fast cell marker gene identification with COSG
Code: https://github.com/genecell/COSG
method: cosg | groupby: leiden | n_groups: 10 | n_genes: 50
**finished identifying marker genes by COSG**
✅ Done | 10 groups × 50 genes | stored in adata.uns['leiden_cosg']
CPU times: user 813 ms, sys: 4.63 ms, total: 817 ms
Wall time: 839 ms
group
rank
names
scores
logfoldchanges
pvals
pvals_adj
pct_group
pct_rest
0
0
1
LYZ
30.139215
8.308955
1.485183e-199
2.036780e-195
1.000000
0.536060
1
0
2
S100A9
29.859339
10.341062
6.640705e-196
4.553531e-192
0.997416
0.245822
2
0
3
S100A8
28.556858
10.218678
2.308892e-179
1.055472e-175
0.968992
0.156552
3
0
4
FCN1
28.287178
8.907052
4.969369e-176
1.703748e-172
0.976744
0.176781
4
0
5
TYROBP
28.184223
8.633690
9.127943e-175
2.503612e-171
1.000000
0.292436
...
...
...
...
...
...
...
...
...
...
195
9
16
RPS27
3.653990
0.227837
2.581968e-04
1.744291e-02
0.984252
0.994475
196
9
17
MT-CYB
3.345087
-0.051800
8.225686e-04
4.283594e-02
0.874016
0.935280
197
9
18
RPS3
3.297153
0.106617
9.767012e-04
4.870720e-02
0.984252
0.995264
198
9
19
RPS16
3.260641
0.137249
1.111605e-03
5.405871e-02
0.976378
0.985793
199
9
20
RPL31
3.222946
0.402909
1.268796e-03
5.918458e-02
0.984252
0.971192
200 rows × 9 columns