Identify the driver regulators of cell fate decisions¶
CEFCON is a computational tool for deciphering driver regulators of cell fate decisions from single-cell RNA-seq data. It takes a prior gene interaction network and expression profiles from scRNA-seq data associated with a given developmental trajectory as inputs, and consists of three main components, including cell-lineage-specific gene regulatory network (GRN) construction, driver regulator identification and regulon-like gene module (RGM) identification.
Check out (Wang et al., Nature Communications, 2023) for the detailed methods and applications.
Code: https://github.com/WPZgithub/CEFCON
import omicverse as ov
#print(f"omicverse version: {ov.__version__}")
import scanpy as sc
#print(f"scanpy version: {sc.__version__}")
import pandas as pd
from tqdm.auto import tqdm
ov.plot_set()
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
Version: 1.5.6, Tutorials: https://omicverse.readthedocs.io/
Data loading and processing¶
Here, we use the mouse hematopoiesis data provided by Nestorowa et al. (2016, Blood).
The scRNA-seq data requires processing to extract lineage information for the CEFCON analysis. Please refer to the original notebook for detailed instructions on preprocessing scRNA-seq data.
adata = ov.single.mouse_hsc_nestorowa16()
adata
Load mouse_hsc_nestorowa16_v0.h5ad
AnnData object with n_obs × n_vars = 1645 × 3000
obs: 'E_pseudotime', 'GM_pseudotime', 'L_pseudotime', 'label_info', 'n_genes', 'leiden', 'cell_type_roughly', 'cell_type_finely'
var: 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'E_pseudotime_logFC', 'GM_pseudotime_logFC', 'L_pseudotime_logFC'
uns: 'cell_type_finely_colors', 'cell_type_roughly_colors', 'draw_graph', 'hvg', 'leiden', 'leiden_colors', 'lineages', 'neighbors', 'pca', 'tsne', 'umap'
obsm: 'X_draw_graph_fa', 'X_pca'
varm: 'PCs'
layers: 'raw_count'
obsp: 'connectivities', 'distances'
CEFCON fully exploit an available global and context-free gene interaction network as prior knowledge, from which we extract the cell-lineage-specific gene interactions according to the gene expression profiles derived from scRNA-seq data associated with a given developmental trajectory.
You can download the prior network in the zenodo. CEFCON only provides the prior network for human and mosue data anaylsis. For other species, you should provide the prior network mannully.
The author of CEFCON has provided several prior networks here; however, ‘nichenet’ yields the best results.
prior_network = ov.single.load_human_prior_interaction_network(dataset='nichenet')
Load the prior gene interaction network: nichenet. #Genes: 25345, #Edges: 5290993
In the scRNA-seq analysis of human data, you should not run this step. Running it may change the gene symbol and result in errors.
# Convert the gene symbols of the prior gene interaction network to the mouse gene symbols
prior_network = ov.single.convert_human_to_mouse_network(prior_network,server_name='asia')
prior_network
Convert genes of the prior interaction network to mouse gene symbols:
Server 'http://asia.ensembl.org/biomart/' is OK
The converted prior gene interaction network: #Genes: 18579, #Edges: 5029532
| from | to | |
|---|---|---|
| 0 | Klf2 | Dlgap1 |
| 2 | Klf2 | Bhlhe40 |
| 3 | Klf2 | Rps6ka1 |
| 4 | Klf2 | Pxn |
| 5 | Klf2 | Ube2v1 |
| ... | ... | ... |
| 837982 | Zranb1 | Zfp141 |
| 837983 | Zranb1 | Zfy1 |
| 837984 | Zranb1 | Zfy2 |
| 837987 | Zscan21 | Zfy1 |
| 837988 | Zscan21 | Zfy2 |
5029532 rows × 2 columns
prior_network.to_csv('result/combined_network_Mouse.txt.gz',sep='\t')
Alternatively, you can directly specify the file path of the input prior interaction network and import the specified file.
#prior_network = './Reference_Networks/combined_network_Mouse.txt'
prior_network=ov.read('result/combined_network_Mouse.txt.gz',index_col=0)
Training CEFCON model¶
We recommend using GRUOBI to solve the integer linear programming (ILP) problem when identifying driver genes. GUROBI is a commercial solver that requires licenses to run. Thankfully, it provides free licenses in academia, as well as trial licenses outside academia. If there is no problem about the licenses, you need to install the gurobipy package.
If difficulties arise while using GUROBI, the non-commercial solver, SCIP, will be employed as an alternative. But the use of SCIP does not come with a guarantee of achieving a successful solutio
By default, the program will verify the availability of GRUOBI. If GRUOBI is not accessible, it will automatically switch the solver to SCIP.
CEFCON_obj = ov.single.pyCEFCON(adata, prior_network, repeats=5, solver='GUROBI')
CEFCON_obj
<omicverse.single._cefcon.pyCEFCON at 0x7f7214e73c40>
Construct cell-lineage-specific GRNs
CEFCON_obj.preprocess()
Start data preparation
[0] - Data loading and preprocessing...
Consider the input data with 3 lineages:
Lineage - E_pseudotime:
2935 extra edges (Spearman correlation > 0.6) are added into the prior gene interaction network.
Total number of edges: 90036.
n_genes × n_cells = 2803 × 1065
Lineage - GM_pseudotime:
100 extra edges (Spearman correlation > 0.6) are added into the prior gene interaction network.
Total number of edges: 90136.
n_genes × n_cells = 2803 × 882
Lineage - L_pseudotime:
4 extra edges (Spearman correlation > 0.6) are added into the prior gene interaction network.
Total number of edges: 90140.
n_genes × n_cells = 2803 × 843
Lineage-by-lineage computation:
CEFCON_obj.train()
Start model training
[1] - Constructing cell-lineage-specific GRN...
Lineage - E_pseudotime:
[1] - Constructing cell-lineage-specific GRN...
Lineage - GM_pseudotime:
[1] - Constructing cell-lineage-specific GRN...
Lineage - L_pseudotime:
Finish model training
# Idenytify driver regulators for each lineage
CEFCON_obj.predicted_driver_regulators()
Start predict lineage - E_pseudotime:
Start calculate gene influence score - E_pseudotime:
Start calculate gene driver regulators - E_pseudotime:
[2] - Identifying driver regulators...
Solving MFVS problem...
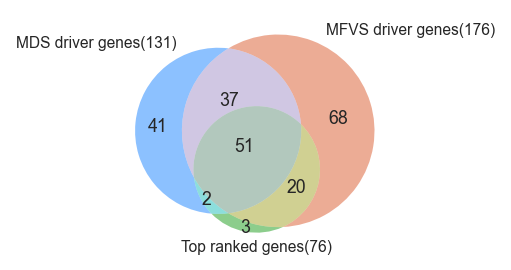
176 critical nodes are found.
0 nodes left after graph reduction operation.
176 MFVS driver genes are found.
Solving MDS problem...
15 critical nodes are found.
1124 nodes left after graph reduction operation.
Solving the Integer Linear Programming problem on the reduced graph...
Solving by GUROBI...(optimal value with GUROBI:116.0, status:optimal)
131 MDS driver genes are found.
Start predict lineage - GM_pseudotime:
Start calculate gene influence score - GM_pseudotime:
Start calculate gene driver regulators - GM_pseudotime:
[2] - Identifying driver regulators...
Solving MFVS problem...
232 critical nodes are found.
0 nodes left after graph reduction operation.
232 MFVS driver genes are found.
Solving MDS problem...
18 critical nodes are found.
1572 nodes left after graph reduction operation.
Solving the Integer Linear Programming problem on the reduced graph...
Solving by GUROBI...(optimal value with GUROBI:206.0, status:optimal)
224 MDS driver genes are found.
Start predict lineage - L_pseudotime:
Start calculate gene influence score - L_pseudotime:
Start calculate gene driver regulators - L_pseudotime:
[2] - Identifying driver regulators...
Solving MFVS problem...
180 critical nodes are found.
4 nodes left after graph reduction operation.
Solving the Integer Linear Programming problem on the reduced graph...
Solving by GUROBI...(optimal value with GUROBI:2.0, status:optimal)
182 MFVS driver nodes are found.
Solving MDS problem...
18 critical nodes are found.
1747 nodes left after graph reduction operation.
Solving the Integer Linear Programming problem on the reduced graph...
Solving by GUROBI...(optimal value with GUROBI:300.0, status:optimal)
318 MDS driver genes are found.
We can find out the driver regulators identified by CEFCON.
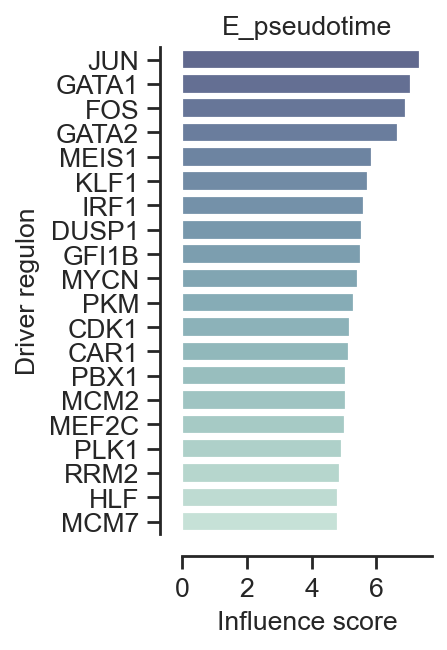
CEFCON_obj.cefcon_results_dict['E_pseudotime'].driver_regulator.head()
| influence_score | is_driver_regulator | is_MFVS_driver | is_MDS_driver | is_TF | |
|---|---|---|---|---|---|
| JUN | 7.352254 | True | True | True | True |
| GATA1 | 7.071392 | True | True | True | True |
| FOS | 6.930125 | True | True | True | True |
| GATA2 | 6.683559 | True | False | True | True |
| MEIS1 | 5.851068 | True | True | True | True |
CEFCON_obj.predicted_RGM()
Start calculate regulon-like gene modules - E_pseudotime:
[3] - Identifying regulon-like gene modules...
Done!
Start calculate regulon-like gene modules - GM_pseudotime:
[3] - Identifying regulon-like gene modules...
Done!
Start calculate regulon-like gene modules - L_pseudotime:
[3] - Identifying regulon-like gene modules...
Done!
Finish predicted
Downstream analysis¶
CEFCON_obj.cefcon_results_dict['E_pseudotime']
CefconResults object with n_cells * n_genes = 1065 * 2748, n_edges = 22424
name: E_pseudotime
expression_data: yes
network: DiGraph with 2748 nodes and 22399 edges
gene_embedding: # dimension = 64
influence_score: yes
driver_regulator: yes
gene_cluster: None
RGMs_AUCell_dict: yes
lineage = 'E_pseudotime'
result = CEFCON_obj.cefcon_results_dict[lineage]
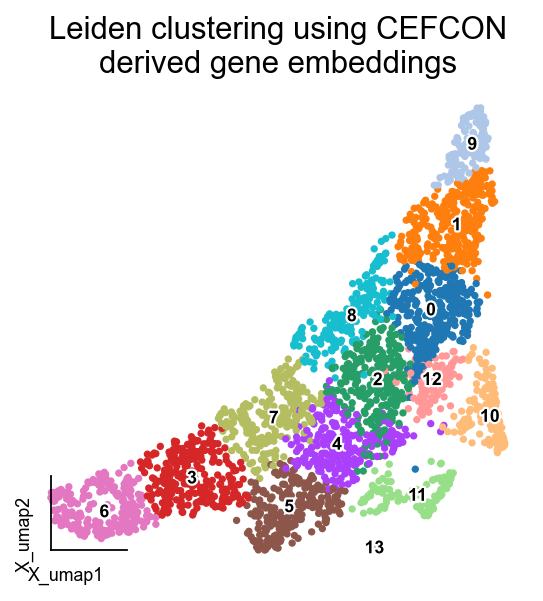
Plot gene embedding clusters
gene_ad=sc.AnnData(result.gene_embedding)
sc.pp.neighbors(gene_ad, n_neighbors=30, use_rep='X')
# Higher resolutions lead to more communities, while lower resolutions lead to fewer communities.
sc.tl.leiden(gene_ad, resolution=1)
sc.tl.umap(gene_ad, n_components=2, min_dist=0.3)
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:01)
running Leiden clustering
finished: found 14 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:01)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:06)
ov.utils.embedding(gene_ad,basis='X_umap',legend_loc='on data',
legend_fontsize=8, legend_fontoutline=2,
color='leiden',frameon='small',title='Leiden clustering using CEFCON\nderived gene embeddings')
Plot influence scores of driver regulators
import matplotlib.pyplot as plt
import seaborn as sns
data_for_plot = result.driver_regulator[result.driver_regulator['is_driver_regulator']]
data_for_plot = data_for_plot[0:20]
plt.figure(figsize=(2, 20 * 0.2))
sns.set_theme(style='ticks', font_scale=0.5)
ax = sns.barplot(x='influence_score', y=data_for_plot.index, data=data_for_plot, orient='h',
palette=sns.color_palette(f"ch:start=.5,rot=-.5,reverse=1,dark=0.4", n_colors=20))
ax.set_title(result.name)
ax.set_xlabel('Influence score')
ax.set_ylabel('Driver regulators')
ax.spines['left'].set_position(('outward', 10))
ax.spines['bottom'].set_position(('outward', 10))
plt.xticks(fontsize=12)
plt.yticks(fontsize=12)
plt.grid(False)
#设置spines可视化情况
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.spines['bottom'].set_visible(True)
ax.spines['left'].set_visible(True)
plt.title('E_pseudotime',fontsize=12)
plt.xlabel('Influence score',fontsize=12)
plt.ylabel('Driver regulon',fontsize=12)
sns.despine()
result.plot_driver_genes_Venn()
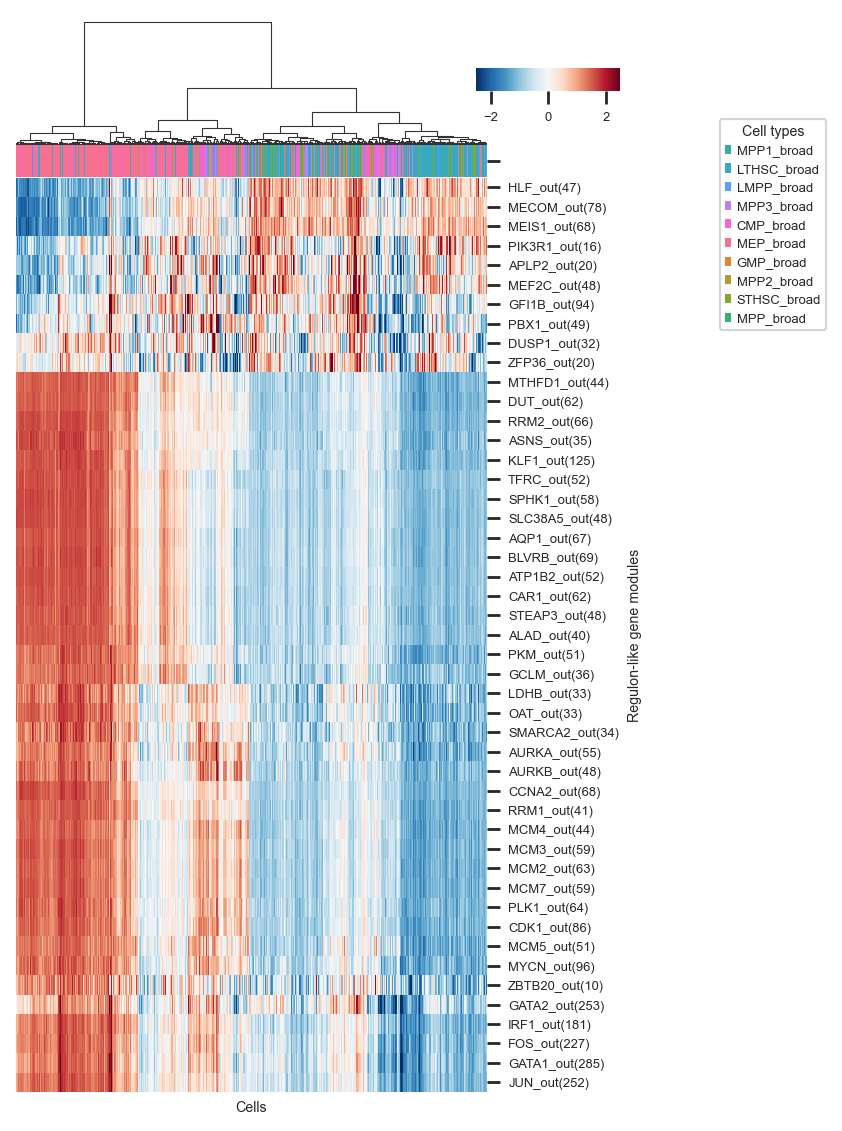
Plot heat map of the activity matrix of RGMs
adata_lineage = adata[adata.obs_names[adata.obs[result.name].notna()],:]
result.plot_RGM_activity_heatmap(cell_label=adata_lineage.obs['cell_type_finely'],
type='out',col_cluster=True,bbox_to_anchor=(1.48, 0.25))
<Figure size 320x320 with 0 Axes>