Bulk RNA-seq mapping with kb-python¶
This notebook demonstrates an alignment-free bulk RNA-seq workflow in OmicVerse using kb-python (kallisto | bustools), starting from SRA files and ending with differential expression (DE) and visualization.
Pipeline overview
Import OmicVerse and set plotting style
Download SRA data (direct
.lite.1links)Convert SRA → paired FASTQ (
parallel_fastq_dump)Download reference genome/annotation (Ensembl GRCh38)
Build kb reference (index + transcript-to-gene map)
Quantify reads with kb-python (
ov.alignment.count, technology=BULK)Merge samples into a single matrix and run DESeq2 via
ov.bulk.pyDEGVisualize DEGs (volcano)
▲ CRITICAL This notebook does not modify any existing code or outputs; it only adds tutorial-style Markdown explanations around the provided cells.
Step 0 — Import OmicVerse¶
We start by importing OmicVerse and setting a consistent plotting style for downstream figures.
%%time
import omicverse as ov
ov.style(font_path='Arial')
🔬 Starting plot initialization...
Using already downloaded Arial font from: /tmp/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] Tesla P40
Memory: 22.4 GB | Compute: 6.1
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 1.7.9rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
CPU times: user 7.88 s, sys: 2.53 s, total: 10.4 s
Wall time: 15.3 s
Step 1 — Download SRA inputs (direct .lite.1 links)¶
Here we download several example SRA runs. In this workflow, we use direct NCBI SRA download links ending with .lite.1.
▲ CRITICAL
ov.datasets.download_data(..., dir=...)will skip downloads if the file already exists.In many environments, you can also download via
ov.alignment.prefetch()and then convert usingfastq-dump/fasterq-dump.If your network blocks direct SRA links, switch to
prefetchor mirror the files to a reachable location.
links=[
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-922/SRR012/12544/SRR12544419/SRR12544419.lite.1',#no
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-924/SRR012/12544/SRR12544421/SRR12544421.lite.1',#no
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-922/SRR012/12544/SRR12544433/SRR12544433.lite.1'#yes
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-922/SRR012/12544/SRR12544435/SRR12544435.lite.1'#yes
]
for link in links:
ov.datasets.download_data(link,dir='./data')
🔍 Downloading data to ./data/SRR12544433.lite.1
✅ Download completed
Step 2 — Convert SRA → paired FASTQ (single example)¶
This cell shows converting one SRA file into paired FASTQ using ov.alignment.parallel_fastq_dump.
▲ CRITICAL
Set
split_files=Truefor paired-end output (*_1.fastq.gz,*_2.fastq.gz).Use a fast local disk for
tmpdirto reduce I/O overhead.Thread count controls conversion speed; choose based on your CPU allocation.
ov.alignment.parallel_fastq_dump(
sra_id='./data/SRR12544421.lite.1',
threads=12,
outdir='./data/SRR12544421',
tmpdir='./tmp',
split_files=True,
gzip=True,
)
🚀 Starting parallel-fastq-dump for ./data/SRR12544421.lite.1
Threads: 12
Output directory: ./data/SRR12544421
Added to PATH: /home/groups/xiaojie/steorra/env/omicverse/bin
>> /home/groups/xiaojie/steorra/env/omicverse/bin/parallel-fastq-dump --sra-id ./data/SRR12544421.lite.1 --threads 12 --outdir ./data/SRR12544421 --tmpdir ./tmp --minSpotId 1 --split-files --gzip
2026-01-29 23:56:27,879 - SRR ids: ['./data/SRR12544421.lite.1']
2026-01-29 23:56:27,879 - extra args: ['--split-files', '--gzip']
2026-01-29 23:56:27,880 - tempdir: ./tmp/pfd_0armgw5l
2026-01-29 23:56:27,880 - CMD: sra-stat --meta --quick ./data/SRR12544421.lite.1
2026-01-29 23:56:27,982 - ./data/SRR12544421.lite.1 spots: 10963094
2026-01-29 23:56:27,982 - blocks: [[1, 913591], [913592, 1827182], [1827183, 2740773], [2740774, 3654364], [3654365, 4567955], [4567956, 5481546], [5481547, 6395137], [6395138, 7308728], [7308729, 8222319], [8222320, 9135910], [9135911, 10049501], [10049502, 10963094]]
2026-01-29 23:56:27,982 - CMD: fastq-dump -N 1 -X 913591 -O ./tmp/pfd_0armgw5l/0 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,985 - CMD: fastq-dump -N 913592 -X 1827182 -O ./tmp/pfd_0armgw5l/1 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,986 - CMD: fastq-dump -N 1827183 -X 2740773 -O ./tmp/pfd_0armgw5l/2 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,987 - CMD: fastq-dump -N 2740774 -X 3654364 -O ./tmp/pfd_0armgw5l/3 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,989 - CMD: fastq-dump -N 3654365 -X 4567955 -O ./tmp/pfd_0armgw5l/4 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,991 - CMD: fastq-dump -N 4567956 -X 5481546 -O ./tmp/pfd_0armgw5l/5 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,992 - CMD: fastq-dump -N 5481547 -X 6395137 -O ./tmp/pfd_0armgw5l/6 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,994 - CMD: fastq-dump -N 6395138 -X 7308728 -O ./tmp/pfd_0armgw5l/7 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,996 - CMD: fastq-dump -N 7308729 -X 8222319 -O ./tmp/pfd_0armgw5l/8 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,998 - CMD: fastq-dump -N 8222320 -X 9135910 -O ./tmp/pfd_0armgw5l/9 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:28,001 - CMD: fastq-dump -N 9135911 -X 10049501 -O ./tmp/pfd_0armgw5l/10 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:28,003 - CMD: fastq-dump -N 10049502 -X 10963094 -O ./tmp/pfd_0armgw5l/11 --split-files --gzip ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913593 spots for ./data/SRR12544421.lite.1
Written 913593 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
✓ parallel-fastq-dump completed successfully!
{'sra_id': './data/SRR12544421.lite.1',
'threads': 12,
'outdir': './data/SRR12544421',
'split_files': True,
'gzip': True}
Step 2b — Batch conversion for multiple SRAs¶
This loop converts multiple SRA files to paired FASTQs using the same settings.
Tip: If you have many samples, consider using a job scheduler (e.g., Slurm) to parallelize across nodes rather than increasing threads indefinitely on one node.
for sra in [
'SRR12544433','SRR12544435'
]:
ov.alignment.parallel_fastq_dump(
sra_id=f'./data/{sra}.lite.1',
threads=12,
outdir=f'./data/{sra}',
tmpdir='./tmp',
split_files=True,
gzip=True,
)
🚀 Starting parallel-fastq-dump for ./data/SRR12544433.lite.1
Threads: 12
Output directory: ./data/SRR12544433
Added to PATH: /home/groups/xiaojie/steorra/env/omicverse/bin
>> /home/groups/xiaojie/steorra/env/omicverse/bin/parallel-fastq-dump --sra-id ./data/SRR12544433.lite.1 --threads 12 --outdir ./data/SRR12544433 --tmpdir ./tmp --minSpotId 1 --split-files --gzip
2026-01-29 23:58:48,245 - SRR ids: ['./data/SRR12544433.lite.1']
2026-01-29 23:58:48,245 - extra args: ['--split-files', '--gzip']
2026-01-29 23:58:48,246 - tempdir: ./tmp/pfd_8tgtda45
2026-01-29 23:58:48,246 - CMD: sra-stat --meta --quick ./data/SRR12544433.lite.1
2026-01-29 23:58:48,358 - ./data/SRR12544433.lite.1 spots: 16602881
2026-01-29 23:58:48,358 - blocks: [[1, 1383573], [1383574, 2767146], [2767147, 4150719], [4150720, 5534292], [5534293, 6917865], [6917866, 8301438], [8301439, 9685011], [9685012, 11068584], [11068585, 12452157], [12452158, 13835730], [13835731, 15219303], [15219304, 16602881]]
2026-01-29 23:58:48,359 - CMD: fastq-dump -N 1 -X 1383573 -O ./tmp/pfd_8tgtda45/0 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,361 - CMD: fastq-dump -N 1383574 -X 2767146 -O ./tmp/pfd_8tgtda45/1 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,362 - CMD: fastq-dump -N 2767147 -X 4150719 -O ./tmp/pfd_8tgtda45/2 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,364 - CMD: fastq-dump -N 4150720 -X 5534292 -O ./tmp/pfd_8tgtda45/3 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,365 - CMD: fastq-dump -N 5534293 -X 6917865 -O ./tmp/pfd_8tgtda45/4 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,367 - CMD: fastq-dump -N 6917866 -X 8301438 -O ./tmp/pfd_8tgtda45/5 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,368 - CMD: fastq-dump -N 8301439 -X 9685011 -O ./tmp/pfd_8tgtda45/6 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,370 - CMD: fastq-dump -N 9685012 -X 11068584 -O ./tmp/pfd_8tgtda45/7 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,372 - CMD: fastq-dump -N 11068585 -X 12452157 -O ./tmp/pfd_8tgtda45/8 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,373 - CMD: fastq-dump -N 12452158 -X 13835730 -O ./tmp/pfd_8tgtda45/9 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,375 - CMD: fastq-dump -N 13835731 -X 15219303 -O ./tmp/pfd_8tgtda45/10 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,377 - CMD: fastq-dump -N 15219304 -X 16602881 -O ./tmp/pfd_8tgtda45/11 --split-files --gzip ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383578 spots for ./data/SRR12544433.lite.1
Written 1383578 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
✓ parallel-fastq-dump completed successfully!
🚀 Starting parallel-fastq-dump for ./data/SRR12544435.lite.1
Threads: 12
Output directory: ./data/SRR12544435
Added to PATH: /home/groups/xiaojie/steorra/env/omicverse/bin
>> /home/groups/xiaojie/steorra/env/omicverse/bin/parallel-fastq-dump --sra-id ./data/SRR12544435.lite.1 --threads 12 --outdir ./data/SRR12544435 --tmpdir ./tmp --minSpotId 1 --split-files --gzip
2026-01-29 23:59:23,325 - SRR ids: ['./data/SRR12544435.lite.1']
2026-01-29 23:59:23,325 - extra args: ['--split-files', '--gzip']
2026-01-29 23:59:23,326 - tempdir: ./tmp/pfd_t7s3okn4
2026-01-29 23:59:23,326 - CMD: sra-stat --meta --quick ./data/SRR12544435.lite.1
2026-01-29 23:59:23,433 - ./data/SRR12544435.lite.1 spots: 16486139
2026-01-29 23:59:23,433 - blocks: [[1, 1373844], [1373845, 2747688], [2747689, 4121532], [4121533, 5495376], [5495377, 6869220], [6869221, 8243064], [8243065, 9616908], [9616909, 10990752], [10990753, 12364596], [12364597, 13738440], [13738441, 15112284], [15112285, 16486139]]
2026-01-29 23:59:23,433 - CMD: fastq-dump -N 1 -X 1373844 -O ./tmp/pfd_t7s3okn4/0 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,435 - CMD: fastq-dump -N 1373845 -X 2747688 -O ./tmp/pfd_t7s3okn4/1 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,436 - CMD: fastq-dump -N 2747689 -X 4121532 -O ./tmp/pfd_t7s3okn4/2 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,438 - CMD: fastq-dump -N 4121533 -X 5495376 -O ./tmp/pfd_t7s3okn4/3 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,439 - CMD: fastq-dump -N 5495377 -X 6869220 -O ./tmp/pfd_t7s3okn4/4 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,441 - CMD: fastq-dump -N 6869221 -X 8243064 -O ./tmp/pfd_t7s3okn4/5 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,443 - CMD: fastq-dump -N 8243065 -X 9616908 -O ./tmp/pfd_t7s3okn4/6 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,445 - CMD: fastq-dump -N 9616909 -X 10990752 -O ./tmp/pfd_t7s3okn4/7 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,451 - CMD: fastq-dump -N 10990753 -X 12364596 -O ./tmp/pfd_t7s3okn4/8 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,453 - CMD: fastq-dump -N 12364597 -X 13738440 -O ./tmp/pfd_t7s3okn4/9 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,455 - CMD: fastq-dump -N 13738441 -X 15112284 -O ./tmp/pfd_t7s3okn4/10 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,456 - CMD: fastq-dump -N 15112285 -X 16486139 -O ./tmp/pfd_t7s3okn4/11 --split-files --gzip ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373855 spots for ./data/SRR12544435.lite.1
Written 1373855 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
✓ parallel-fastq-dump completed successfully!
Step 3 — Download reference genome + annotation¶
We download GRCh38 reference FASTA and GTF from Ensembl.
▲ CRITICAL
Keep the FASTA and GTF from the same Ensembl release to avoid mismatches in transcript IDs.
Ensure enough disk space (human reference files are large).
ov.datasets.download_data('ftp://ftp.ensembl.org/pub/release-108/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz',
dir='./genomes')
ov.datasets.download_data('ftp://ftp.ensembl.org/pub/release-108/gtf/homo_sapiens/Homo_sapiens.GRCh38.108.gtf.gz',
dir='./genomes')
🔍 Downloading data to ./genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
✅ Download completed
🔍 Downloading data to ./genomes/Homo_sapiens.GRCh38.108.gtf.gz
✅ Download completed
'./genomes/Homo_sapiens.GRCh38.108.gtf.gz'
Step 4 — Build kb-python reference (index + t2g + cDNA)¶
ov.alignment.single.ref(...) prepares the kb-python reference assets:
index.idx: kallisto indext2g.txt: transcript-to-gene mapping used to aggregate counts to genescdna.fa: transcriptome FASTA derived from the annotation
▲ CRITICAL
The first build can be time-consuming; once built, reuse the same outputs for all samples.
Place the index on fast storage to speed up quantification.
result = ov.alignment.single.ref(
fasta_paths='genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz', #input
gtf_paths='genomes/Homo_sapiens.GRCh38.108.gtf.gz', #input
index_path='pbmc_1k_v3/index.idx', #output
t2g_path='pbmc_1k_v3/t2g.txt', #output
cdna_path='pbmc_1k_v3/cdna.fa', #output
temp_dir='tmp',
overwrite=True,
)
print(result.keys())
🚀 Starting ref workflow: standard
Using temporary directory: tmp-kb-e9513f2c23aa46d1961cde1d7afeba6b
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb ref --tmp tmp-kb-e9513f2c23aa46d1961cde1d7afeba6b -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -t 8 --overwrite --d-list-overhang 1 -f1 pbmc_1k_v3/cdna.fa pbmc_1k_v3/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz pbmc_1k_v3/Homo_sapiens.GRCh38.108.gtf.gz
[2026-01-29 22:21:41,856] INFO [ref] Preparing pbmc_1k_v3/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz, pbmc_1k_v3/Homo_sapiens.GRCh38.108.gtf.gz
[2026-01-29 22:22:33,317] INFO [ref] Splitting genome pbmc_1k_v3/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz into cDNA at /scratch/users/steorra/analysis/26_omic_protocol/tmp-kb-e9513f2c23aa46d1961cde1d7afeba6b/tmpoyd3o7ta
[2026-01-29 22:23:30,907] INFO [ref] Concatenating 1 cDNAs to pbmc_1k_v3/cdna.fa
[2026-01-29 22:23:31,551] INFO [ref] Creating transcript-to-gene mapping at pbmc_1k_v3/t2g.txt
[2026-01-29 22:23:33,230] INFO [ref] Indexing pbmc_1k_v3/cdna.fa to pbmc_1k_v3/index.idx
✓ ref workflow completed!
dict_keys(['workflow', 'technology', 'parameters', 'index_path', 't2g_path', 'cdna_path'])
Step 5 — Quantify one sample with kb-python (technology = BULK)¶
This is an example call for one sample.
▲ CRITICAL
For bulk RNA-seq, each sample is treated as one “library” (no barcode filtering).
h5ad=Truewrites an AnnData object for convenient downstream analysis in OmicVerse/Scanpy.If your environment does not expose
count(...)directly, the equivalent OmicVerse entry point is typicallyov.alignment.count(...)(the rest of the arguments stay the same).
result = count(
fastq_paths=[
"./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz",
"./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz",
],
index_path="pbmc_1k_v3/index.idx",
t2g_path="pbmc_1k_v3/t2g.txt",
technology='BULK', # technology
output_path="results/pbmc_test",
h5ad=True,
filter_barcodes=False,
threads=12,
parity="paired", #
strand="unstranded", #
)
print(result.keys())
🚀 Starting count workflow: standard
Technology: BULK
Output directory: .
Using temporary directory: tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2 -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o . -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-29 22:53:46,113] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to . from
[2026-01-29 22:53:46,113] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz
[2026-01-29 22:53:46,113] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-29 22:54:23,070] INFO [count] Sorting BUS file ./output.bus to tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2/output.s.bus
[2026-01-29 22:54:25,476] INFO [count] Inspecting BUS file tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2/output.s.bus
[2026-01-29 22:54:26,580] INFO [count] Generating count matrix ./counts_unfiltered/cells_x_genes from BUS file tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2/output.s.bus
[2026-01-29 22:54:28,602] INFO [count] Writing gene names to file ./counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-29 22:54:28,835] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-29 22:54:28,860] INFO [count] Reading matrix ./counts_unfiltered/cells_x_genes.mtx
[2026-01-29 22:54:28,917] INFO [count] Writing matrix to h5ad ./counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
Step 5b — Quantify all samples (loop)¶
We run kb-python quantification for each sample and save results into per-sample output folders.
Tip: In practice, you may want to set a clear naming convention for output_path (project/sample/date) to keep results reproducible.
for sra in [
'SRR12544419','SRR12544421','SRR12544433','SRR12544435'
]:
result = ov.alignment.count(
fastq_paths=[
f"./data/{sra}/{sra}.lite.1_1.fastq.gz",
f"./data/{sra}/{sra}.lite.1_2.fastq.gz",
],
index_path="pbmc_1k_v3/index.idx",
t2g_path="pbmc_1k_v3/t2g.txt",
technology='BULK', # technology
output_path=f"results/{sra}/",
h5ad=True,
filter_barcodes=False,
threads=12,
parity="paired", # ✅关键
strand="unstranded", # ✅建议显式写
)
print(result.keys())
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544419/
Using temporary directory: tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544419/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-30 00:03:00,630] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544419/ from
[2026-01-30 00:03:00,630] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz
[2026-01-30 00:03:00,630] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-30 00:03:35,180] INFO [count] Sorting BUS file results/SRR12544419/output.bus to tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc/output.s.bus
[2026-01-30 00:03:37,594] INFO [count] Inspecting BUS file tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc/output.s.bus
[2026-01-30 00:03:38,702] INFO [count] Generating count matrix results/SRR12544419/counts_unfiltered/cells_x_genes from BUS file tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc/output.s.bus
[2026-01-30 00:03:41,325] INFO [count] Writing gene names to file results/SRR12544419/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:03:41,560] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:03:41,586] INFO [count] Reading matrix results/SRR12544419/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:03:41,643] INFO [count] Writing matrix to h5ad results/SRR12544419/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544421/
Using temporary directory: tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544421/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544421/SRR12544421.lite.1_1.fastq.gz ./data/SRR12544421/SRR12544421.lite.1_2.fastq.gz
[2026-01-30 00:03:53,957] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544421/ from
[2026-01-30 00:03:53,957] INFO [count] ./data/SRR12544421/SRR12544421.lite.1_1.fastq.gz
[2026-01-30 00:03:53,957] INFO [count] ./data/SRR12544421/SRR12544421.lite.1_2.fastq.gz
[2026-01-30 00:04:18,493] INFO [count] Sorting BUS file results/SRR12544421/output.bus to tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e/output.s.bus
[2026-01-30 00:04:20,598] INFO [count] Inspecting BUS file tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e/output.s.bus
[2026-01-30 00:04:21,702] INFO [count] Generating count matrix results/SRR12544421/counts_unfiltered/cells_x_genes from BUS file tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e/output.s.bus
[2026-01-30 00:04:23,724] INFO [count] Writing gene names to file results/SRR12544421/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:04:23,956] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:04:23,981] INFO [count] Reading matrix results/SRR12544421/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:04:24,037] INFO [count] Writing matrix to h5ad results/SRR12544421/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544433/
Using temporary directory: tmp-kb-f36592f2f361482c8829daecfd59d0a4
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-f36592f2f361482c8829daecfd59d0a4 -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544433/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544433/SRR12544433.lite.1_1.fastq.gz ./data/SRR12544433/SRR12544433.lite.1_2.fastq.gz
[2026-01-30 00:04:33,656] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544433/ from
[2026-01-30 00:04:33,656] INFO [count] ./data/SRR12544433/SRR12544433.lite.1_1.fastq.gz
[2026-01-30 00:04:33,656] INFO [count] ./data/SRR12544433/SRR12544433.lite.1_2.fastq.gz
[2026-01-30 00:05:04,901] INFO [count] Sorting BUS file results/SRR12544433/output.bus to tmp-kb-f36592f2f361482c8829daecfd59d0a4/output.s.bus
[2026-01-30 00:05:07,209] INFO [count] Inspecting BUS file tmp-kb-f36592f2f361482c8829daecfd59d0a4/output.s.bus
[2026-01-30 00:05:08,313] INFO [count] Generating count matrix results/SRR12544433/counts_unfiltered/cells_x_genes from BUS file tmp-kb-f36592f2f361482c8829daecfd59d0a4/output.s.bus
[2026-01-30 00:05:10,436] INFO [count] Writing gene names to file results/SRR12544433/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:05:10,667] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:05:10,693] INFO [count] Reading matrix results/SRR12544433/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:05:10,749] INFO [count] Writing matrix to h5ad results/SRR12544433/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544435/
Using temporary directory: tmp-kb-7e11e84640b14489ad063057ba8b2738
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-7e11e84640b14489ad063057ba8b2738 -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544435/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544435/SRR12544435.lite.1_1.fastq.gz ./data/SRR12544435/SRR12544435.lite.1_2.fastq.gz
[2026-01-30 00:05:21,328] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544435/ from
[2026-01-30 00:05:21,328] INFO [count] ./data/SRR12544435/SRR12544435.lite.1_1.fastq.gz
[2026-01-30 00:05:21,328] INFO [count] ./data/SRR12544435/SRR12544435.lite.1_2.fastq.gz
[2026-01-30 00:05:52,473] INFO [count] Sorting BUS file results/SRR12544435/output.bus to tmp-kb-7e11e84640b14489ad063057ba8b2738/output.s.bus
[2026-01-30 00:05:54,882] INFO [count] Inspecting BUS file tmp-kb-7e11e84640b14489ad063057ba8b2738/output.s.bus
[2026-01-30 00:05:55,986] INFO [count] Generating count matrix results/SRR12544435/counts_unfiltered/cells_x_genes from BUS file tmp-kb-7e11e84640b14489ad063057ba8b2738/output.s.bus
[2026-01-30 00:05:58,108] INFO [count] Writing gene names to file results/SRR12544435/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:05:58,341] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:05:58,366] INFO [count] Reading matrix results/SRR12544435/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:05:58,424] INFO [count] Writing matrix to h5ad results/SRR12544435/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
Step 6 — Load per-sample results and harmonize gene identifiers¶
Each kb-python run generates an adata.h5ad plus companion gene name files.
We load per-sample AnnData objects and ensure adata.var contains both:
gene_namegene_id
▲ CRITICAL
Gene naming conventions can differ across pipelines; explicitly setting adata.var['gene_name'] and using it as index makes downstream merging and visualization more robust.
ad_dict={}
for sra in [
'SRR12544419','SRR12544421','SRR12544433','SRR12544435'
]:
ad=ov.read(f'./results/{sra}/counts_unfiltered/adata.h5ad')
gene_name=ov.pd.read_csv(
f'./results/{sra}/counts_unfiltered/cells_x_genes.genes.names.txt',
header=None
)
ad.var['gene_name']=gene_name[0].tolist()
ad.var['gene_id']=ad.var.index
ad.var.index=ad.var['gene_name']
ad.var_names_make_unique()
ad.obs['sra']=sra
ad_dict[sra]=ad
Step 7 — Merge samples and define phenotype labels¶
We concatenate all samples into one AnnData and create a Group column (e.g., disease vs healthy) for downstream DE.
▲ CRITICAL
For bulk RNA-seq, you typically want one observation per sample. In this demo workflow, each sample’s output is concatenated and then labeled via adata.obs['Group'].
adata=ov.concat(ad_dict)
adata.obs_names_make_unique()
adata.obs['Group']=['no','no','yes','yes']
adata
AnnData object with n_obs × n_vars = 4 × 62703
obs: 'sra', 'Group'
Step 7b — Quick sanity check (gene expression)¶
A quick check on a marker gene can help confirm that the matrix was loaded correctly and contains non-zero counts where expected.
adata[:,'CD3D'].X.toarray()
array([[623.],
[612.],
[ 98.],
[324.]])
Step 8 — Convert to a count matrix for DE¶
ov.bulk.pyDEG expects a gene × sample count matrix (pandas DataFrame).
Here we convert AnnData to a DataFrame and transpose to match that convention.
▲ CRITICAL DESeq2-style methods require raw integer counts. Do not apply log-normalization before DESeq2.
data=adata.to_df().T
data.head()
| barcode | AAAAAAAAAAAAAAAA | AAAAAAAAAAAAAAAA-1 | AAAAAAAAAAAAAAAA-2 | AAAAAAAAAAAAAAAA-3 |
|---|---|---|---|---|
| gene_name | ||||
| ATAD3B | 90.0 | 70.0 | 30.0 | 115.0 |
| DDX11L17 | 4.0 | 0.0 | 0.0 | 0.0 |
| ENSG00000228037.1 | 9.0 | 8.0 | 1.0 | 8.0 |
| PRDM16 | 0.0 | 0.0 | 0.0 | 0.0 |
| ENSG00000284616.1 | 0.0 | 0.0 | 0.0 | 0.0 |
Step 9 — Create a pyDEG object¶
We wrap the count matrix into an OmicVerse pyDEG object for differential expression analysis.
dds=ov.bulk.pyDEG(data)
Step 10 — Differential expression (DESeq2)¶
We run DE analysis using the DESeq2 backend.
▲ CRITICAL
DESeq2 expects raw integer counts and internally estimates size factors/dispersion.
The
treatment_groupsandcontrol_groupshere refer to the column/sample identifiers in the provided matrix. In your own data, replace them with the appropriate sample IDs (often the sample names).
dds.drop_duplicates_index()
print('... drop_duplicates_index success')
treatment_groups=['AAAAAAAAAAAAAAAA-2','AAAAAAAAAAAAAAAA-3']
control_groups=['AAAAAAAAAAAAAAAA','AAAAAAAAAAAAAAAA-1']
result=dds.deg_analysis(treatment_groups,control_groups,method='DEseq2')
... drop_duplicates_index success
⚙️ You are using DEseq2 method for differential expression analysis.
⏰ Start to create DeseqDataSet...
logres_prior=1.2473932639674563, sigma_prior=0.25
Log2 fold change & Wald test p-value: condition Treatment vs Control
baseMean log2FoldChange lfcSE stat \
gene_name
B2M 178491.812733 0.065096 0.418607 0.155506
S100A9 170557.796728 0.378402 0.248436 1.523133
S100A8 154015.888583 0.914504 0.455013 2.009840
MT-RNR2 143254.671256 0.434606 0.553576 0.785089
HBB 127090.703257 2.661912 1.449787 1.836071
... ... ... ... ...
ENSG00000273937.1 0.000000 NaN NaN NaN
ENSG00000278633.1 0.000000 NaN NaN NaN
ENSG00000278066.1 0.000000 NaN NaN NaN
ENSG00000277374.1 0.000000 NaN NaN NaN
ENSG00000275661.1 0.000000 NaN NaN NaN
pvalue padj
gene_name
B2M 0.876422 0.996897
S100A9 0.127725 0.669610
S100A8 0.044448 0.402458
MT-RNR2 0.432402 0.948016
HBB 0.066347 0.493618
... ... ...
ENSG00000273937.1 NaN NaN
ENSG00000278633.1 NaN NaN
ENSG00000278066.1 NaN NaN
ENSG00000277374.1 NaN NaN
ENSG00000275661.1 NaN NaN
[62703 rows x 6 columns]
✅ Differential expression analysis completed.
Step 11 — Filter low-expression genes¶
Filtering removes genes with insufficient signal and can improve interpretability and multiple-testing behavior.
Here we apply a BaseMean threshold after DE is computed.
print(result.shape)
result=result.loc[result['log2(BaseMean)']>1]
print(result.shape)
(21002, 14)
(21002, 14)
Step 12 — Set DEG thresholds¶
We set thresholds for significance and fold-change, which control DEG calling and downstream visualization.
# -1 means automatically calculates
dds.foldchange_set(fc_threshold=-1,
pval_threshold=0.05,
logp_max=10)
... Fold change threshold: 2.9757964388302165
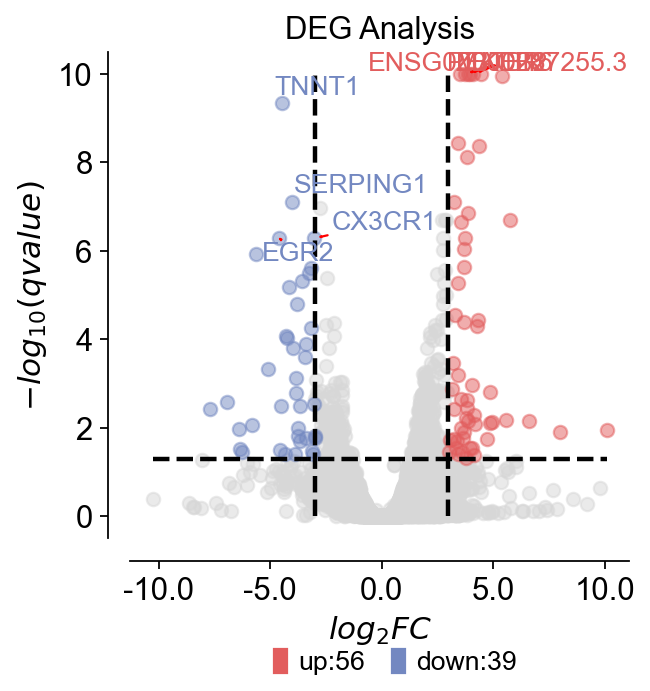
Step 13 — Visualize DEGs (volcano plot)¶
A volcano plot summarizes effect size (log2FC) vs significance.
Tip: After identifying DEGs, typical next steps include:
Gene set enrichment (GO/KEGG/Reactome)
Pathway visualization
PPI network analysis
(Those downstream steps can be added as additional sections depending on your tutorial scope.)
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
plot_genes_num=8,plot_genes_fontsize=12,)
🌋 Volcano Plot Analysis:
Total genes: 62703
↗️ Upregulated genes: 56
↘️ Downregulated genes: 39
➡️ Non-significant genes: 62608
🎯 Total significant genes: 95
log2FC range: -10.26 to 10.10
qvalue range: 5.94e-14 to 1.00e+00
⚙️ Current Function Parameters:
Data columns: pval_name='qvalue', fc_name='log2FC'
Thresholds: pval_threshold=0.05, fc_max=2.9757964388302165, fc_min=-2.9757964388302165
Plot size: figsize=(4, 4)
Gene labels: plot_genes_num=8, plot_genes_fontsize=12
Custom genes: None (auto-select top genes)
💡 Parameter Optimization Suggestions:
✅ Current parameters are optimal for your data!
────────────────────────────────────────────────────────────
<Axes: title={'center': 'DEG Analysis'}, xlabel='$log_{2}FC$', ylabel='$-log_{10}(qvalue)$'>
Notes on scaling and reproducibility¶
CPU & memory: kb-python is usually lighter than full-genome alignment, but index building and quantification still benefit from multiple threads and fast disk.
Reference consistency: Always keep FASTA/GTF release versions consistent (and document the release in your tutorial).
Project structure: Consider organizing outputs as
results/<project>/<sample>/...and saving ametadata.csvdescribing groups/replicates.
If you want, I can extend this notebook by adding new Markdown + new code cells for enrichment and PPI analysis (without touching existing cells), following the same OmicVerse tutorial style.