Differential expression and celltype analysis [All Cell]¶
Differential gene expression (DGE) analysis identifies genes that show statistically significant differences in expression levels across distinct cell populations or conditions. This analysis helps in identifying which cell types are most affected by a condition of interest such as a disease, and characterizing their functional signatures.
Differential Compositional analysis identifies Quantifies changes in the relative abundances of each cell type across conditions (e.g., case vs. control, time points, treatment groups). Reveals expansions or contractions of specific populations that may not be captured by gene-level analyses alone.
Here, we introduced omicverse.single.DEG and omicverse.single.DCT to performed these two analysis.
import scanpy as sc
#import pertpy as pt
import omicverse as ov
ov.style()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ Apple Silicon MPS detected
• [MPS] Apple Silicon GPU - Metal Performance Shaders available
✅ plot_set complete.
Data Preprocess¶
The data we use in the following example comes from [Haber et al., 2017]. It contains samples from the small intestinal epithelium of mice with different conditions. We first load the raw cell-level data. The dataset contains gene expressions of 9842 cells. They are annotated with their sample identifier (batch), the condition of the subjects and the type of each cell (cell_label).
adata = pt.dt.haber_2017_regions()
adata
AnnData object with n_obs × n_vars = 9842 × 15215
obs: 'batch', 'barcode', 'condition', 'cell_label'
For our first example, we want to look at how the Salmonella infection influences the cell composition. Therefore, we create a subset of our compositional data that only contains the healthy and Salmonella-infected samples as a new data modality.
# Select control and salmonella data
adata = adata[
adata.obs["condition"].isin(["Control", "Salmonella"])
].copy()
print(adata)
AnnData object with n_obs × n_vars = 5010 × 15215
obs: 'batch', 'barcode', 'condition', 'cell_label'
adata.obs["condition"].unique()
['Control', 'Salmonella']
Categories (2, object): ['Control', 'Salmonella']
DEG with wilcoxon/t-test¶
In omicverse, we only need one function ov.single.DEG to perfrom all DEG analysis tasks. First, I will introduce the nonparametric Wilcoxon test and the t-test—two widely used methods for differential expression analysis. Due to their high computational efficiency, we applied them in our DEG analysis.
We need to set the ctrl_group and test_group to perform the analysis. Besides, we also need to define the celltype to be explode. If you set celltype_group is None, all the celltype will be calculated.
deg_obj=ov.single.DEG(
adata,
condition='condition',
ctrl_group='Control',
test_group='Salmonella',
method='wilcoxon',
)
deg_obj.run(
celltype_key='cell_label',
celltype_group=['TA'],
)
✅ Differential expression analysis initialized
📊 DEG analysis using wilcoxon method
📊 Condition: condition, Control group: Control, Test group: Salmonella
📊 Celltype key: cell_label, Celltype group: ['TA']
Total cells: 533 will be used for DEG analysis
normalizing counts per cell
finished (0:00:00)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:00)
✅ wilcoxon DEG analysis completed
res_wilcoxon=deg_obj.get_results()
res_wilcoxon.head()
| log2FC | pvalue | padj | qvalue | size | sig | -log(pvalue) | -log(qvalue) | |
|---|---|---|---|---|---|---|---|---|
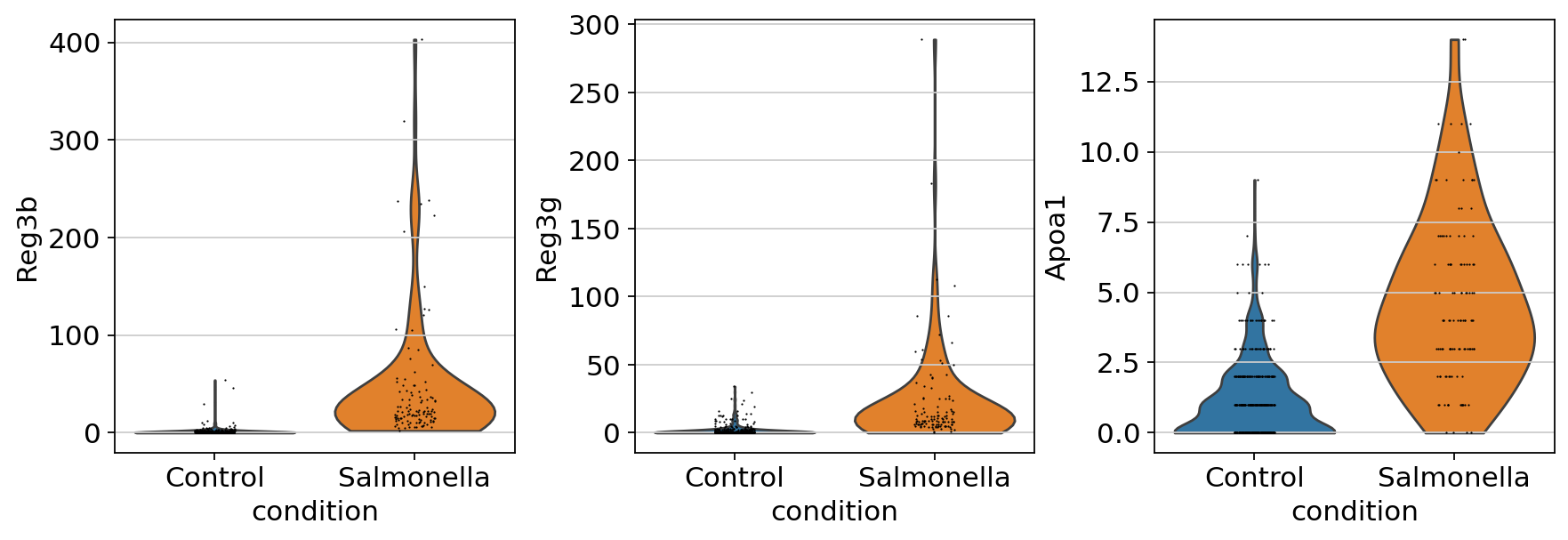
| Reg3b | 5.958403 | 2.899264e-58 | 4.411230e-54 | 4.411230e-54 | 0.595840 | sig | 57.537712 | 53.355440 |
| Reg3g | 4.425004 | 1.576382e-51 | 1.199233e-47 | 1.199233e-47 | 0.442500 | sig | 50.802339 | 46.921097 |
| Apoa1 | 2.589844 | 2.025723e-40 | 1.027379e-36 | 1.027379e-36 | 0.258984 | sig | 39.693420 | 35.988269 |
| Guca2b | 2.380329 | 1.732794e-21 | 5.272893e-18 | 5.272893e-18 | 0.238033 | sig | 20.761253 | 17.277951 |
| Zg16 | 2.077089 | 2.553716e-21 | 6.475798e-18 | 6.475798e-18 | 0.207709 | sig | 20.592827 | 17.188707 |
We can use sc.pl.violin to compare the gene expression between different condition.
celltypes_li=['TA']
sc.pl.violin(
adata[adata.obs['cell_label'].isin(celltypes_li)],
keys=['Reg3b','Reg3g','Apoa1'],
groupby='condition'
)
DEG with memento¶
memento is a python package for performing differential mean, variability, and correlation in single-cell RNA sequencing data.
Memento, an end-to-end method that implements a hierarchical model for estimating mean, residual variance, and gene correlation from scRNA-seq data and provides a statistical framework for hypothesis testing of these parameters.
deg_obj=ov.single.DEG(
adata,
condition='condition',
ctrl_group='Control',
test_group='Salmonella',
method='memento-de',
)
deg_obj.run(
celltype_key='cell_label',
celltype_group=['TA'],
capture_rate=0.07,
num_cpus=12,
num_boot=5000
)
✅ Differential expression analysis initialized
📊 DEG analysis using memento-de method
📊 Condition: condition, Control group: Control, Test group: Salmonella
📊 Celltype key: cell_label, Celltype group: ['TA']
Total cells: 533 will be used for DEG analysis
✅ memento-de DEG analysis completed
res_memento=deg_obj.get_results()
res_memento.query('dv_coef > 1 & de_coef > 0').sort_values('dv_pval').head(5)
| gene | tx | de_coef | de_se | de_pval | dv_coef | dv_se | dv_pval | |
|---|---|---|---|---|---|---|---|---|
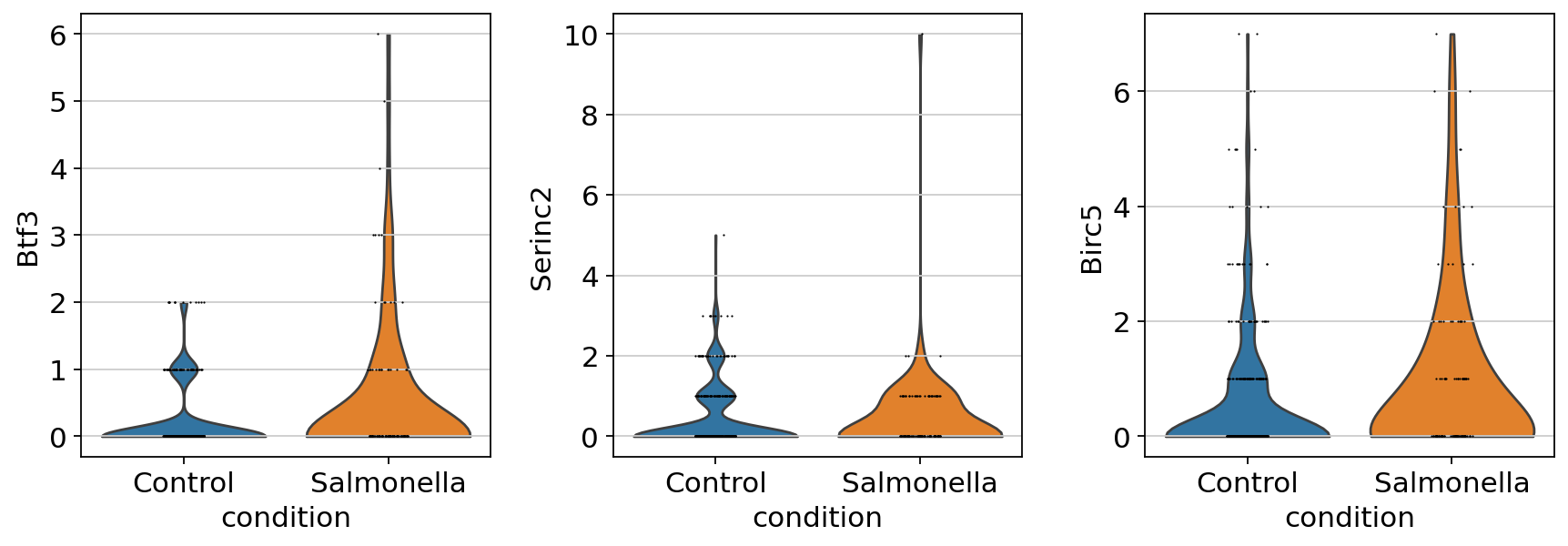
| 443 | Btf3 | stim | 0.838806 | 0.230108 | 0.000213 | 2.940342 | 0.580788 | 4.519054e-07 |
| 397 | Birc5 | stim | 0.631178 | 0.185273 | 0.000661 | 1.198823 | 0.313596 | 8.280704e-05 |
| 3421 | Serinc2 | stim | 0.030935 | 0.238055 | 0.826321 | 2.288608 | 0.511458 | 1.560657e-04 |
| 917 | Dcaf8 | stim | 0.037631 | 0.316794 | 0.846642 | 1.833301 | 0.500972 | 2.687823e-04 |
| 536 | Ccnb2 | stim | 0.132247 | 0.234056 | 0.565606 | 1.126923 | 0.323989 | 3.477457e-04 |
celltypes_li=['TA']
sc.pl.violin(
adata[adata.obs['cell_label'].isin(celltypes_li)],
keys=['Btf3','Serinc2','Birc5'],
groupby='condition'
)
DCT with scCODA¶
In omicverse, we only need one function ov.single.DCT to perfrom all DCT analysis tasks. We included scCODA and milo to perform the celltype abundance analysis.
Besides, you can also perform the analysis with pertpy’s api. dct_obj.model will be helpful.
More tutorial could be found in https://pertpy.readthedocs.io/en/stable/tutorials/notebooks/sccoda.html
dct_obj=ov.single.DCT(
adata,
condition='condition',
ctrl_group='Control',
test_group='Salmonella',
cell_type_key='cell_label',
method='sccoda',
sample_key='batch',
)
• Automatic reference selection! Reference cell type set to Endocrine
No-U-turn HMC sampling is then initiated by calling sccoda_model.run_nuts().
We can use help(dct_obj.model.run_nuts) to obtain the argument as input.
dct_obj.run(
num_samples=5000, #number of sampled values after burn-in.
num_warmup=500, #Number of burn-in (warmup) samples.
)
res=dct_obj.get_results()
res.head()
| Final Parameter | HDI 3% | HDI 97% | SD | Inclusion probability | Expected Sample | log2-fold change | ||
|---|---|---|---|---|---|---|---|---|
| Covariate | Cell Type | |||||||
| conditionT.Salmonella | Endocrine | 0.000000 | 0.000 | 0.000 | 0.000 | 0.0000 | 25.782816 | -0.495162 |
| Enterocyte | 1.367619 | 0.864 | 1.886 | 0.262 | 1.0000 | 325.488661 | 1.477896 | |
| Enterocyte.Progenitor | 0.000000 | -0.393 | 0.647 | 0.163 | 0.3072 | 100.454836 | -0.495162 | |
| Goblet | 0.000000 | -0.228 | 1.037 | 0.298 | 0.4688 | 43.541227 | -0.495162 | |
| Stem | 0.000000 | -0.750 | 0.251 | 0.199 | 0.3560 | 120.266274 | -0.495162 |
Adjusting the False discovery rate¶
scCODA selects credible effects based on their inclusion probability. The cutoff between credible and non-credible effects depends on the desired false discovery rate (FDR). A smaller FDR value will produce more conservative results, but might miss some effects, while a larger FDR value selects more effects at the cost of a larger number of false discoveries.
The desired FDR level can be easily set after inference via sim_results.set_fdr(). Per default, the value is 0.05, but we recommend to increase it up to 0.2 if no effects are found at a more conservative level.
In our example, setting a desired FDR of 0.4 reveals small effects on Endocrine and Tuft cells. Keep in mind that we chose this value only for instructive purposes, since there are no credible effects beside Enterocytes at lower FDR levels. In practice, expecting 40% of all credible effects to be false-positives is usually not recommended.
dct_obj.model.set_fdr(dct_obj.sccoda_data,
modality_key="coda",
est_fdr=0.6)
res=dct_obj.get_results()
res.sort_values('Final Parameter',ascending=False).head()
| Final Parameter | HDI 3% | HDI 97% | SD | Inclusion probability | Expected Sample | log2-fold change | ||
|---|---|---|---|---|---|---|---|---|
| Covariate | Cell Type | |||||||
| conditionT.Salmonella | Enterocyte | 1.367619 | 0.864 | 1.886 | 0.262 | 1.0000 | 327.583710 | 1.487152 |
| Goblet | 0.353104 | -0.228 | 1.037 | 0.298 | 0.4688 | 62.378974 | 0.023516 | |
| Enterocyte.Progenitor | 0.115878 | -0.393 | 0.647 | 0.163 | 0.3072 | 113.522620 | -0.318729 | |
| TA.Early | 0.008501 | -0.408 | 0.486 | 0.121 | 0.2748 | 142.968607 | -0.473642 | |
| Endocrine | 0.000000 | 0.000 | 0.000 | 0.000 | 0.0000 | 25.948771 | -0.485906 |
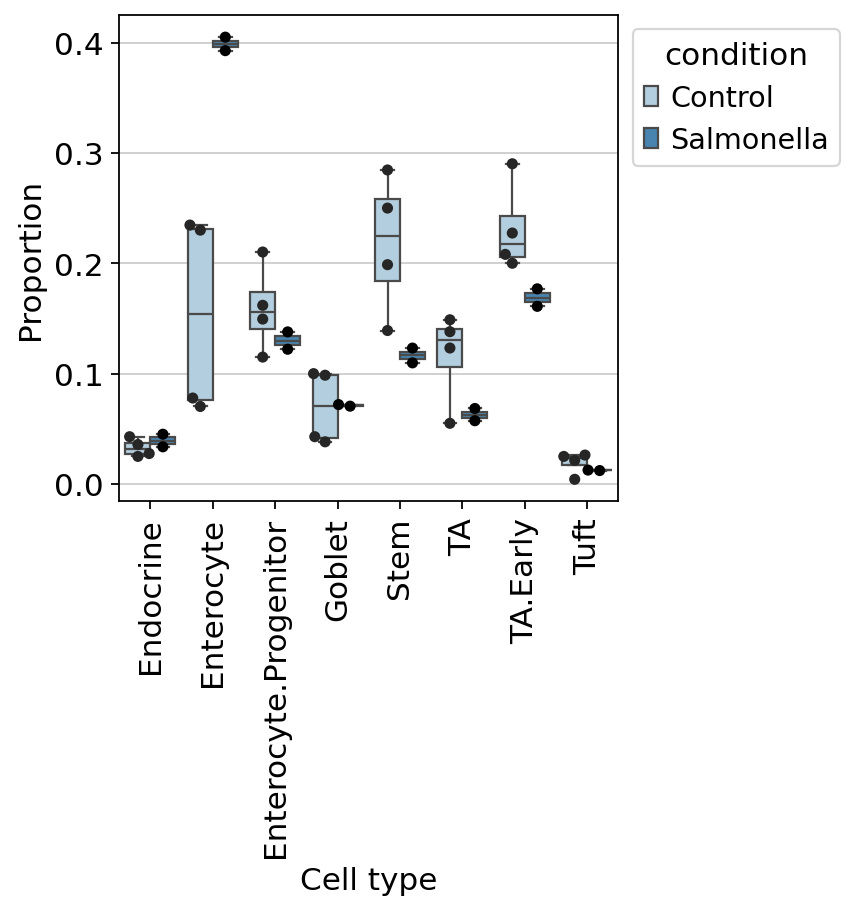
#ov.plot_set()
dct_obj.model.plot_boxplots(
dct_obj.sccoda_data,
modality_key="coda",
feature_name="condition",
add_dots=True,
figsize=(4,4),
dpi=80,
)
ov.plt.show()
dct_obj.model.summary(
dct_obj.sccoda_data,
modality_key="coda"
)
Compositional Analysis summary ┌──────────────────────────────────────────────┬──────────────────────────────────────────────────────────────────┐ │ Name │ Value │ ├──────────────────────────────────────────────┼──────────────────────────────────────────────────────────────────┤ │ Data │ Data: 6 samples, 8 cell types │ │ Reference cell type │ Endocrine │ │ Formula │ condition │ └──────────────────────────────────────────────┴──────────────────────────────────────────────────────────────────┘
┌─────────────────────────────────────────────────────────────────────────────────────────────────────────────────┐ │ Intercepts │ ├─────────────────────────────────────────────────────────────────────────────────────────────────────────────────┤ │ Final Parameter Expected Sample │ │ Cell Type │ │ Endocrine 1.183 36.340 │ │ Enterocyte 2.351 116.854 │ │ Enterocyte.Progenitor 2.543 141.589 │ │ Goblet 1.707 61.370 │ │ Stem 2.723 169.513 │ │ TA 2.126 93.310 │ │ TA.Early 2.881 198.528 │ │ Tuft 0.452 17.495 │ └─────────────────────────────────────────────────────────────────────────────────────────────────────────────────┘
┌─────────────────────────────────────────────────────────────────────────────────────────────────────────────────┐ │ Effects │ ├─────────────────────────────────────────────────────────────────────────────────────────────────────────────────┤ │ Final Parameter Expected Sample log2-fold change │ │ Covariate Cell Type │ │ conditionT.Salmonella Endocrine 0.000 25.949 -0.486 │ │ Enterocyte 1.368 327.584 1.487 │ │ Enterocyte.Progenitor 0.116 113.523 -0.319 │ │ Goblet 0.353 62.379 0.024 │ │ Stem -0.229 96.260 -0.816 │ │ TA -0.212 53.916 -0.791 │ │ TA.Early 0.009 142.969 -0.474 │ │ Tuft -0.006 12.421 -0.494 │ └─────────────────────────────────────────────────────────────────────────────────────────────────────────────────┘
DCT with milopy¶
Many biological conditions (disease, development, genetic KOs) can induce shifts in cell composition, where cells of a given state become enriched or depleted in response to a perturbation. With differential abundance analysis, we quantify consistent changes in cell composition across replicate samples. While differential abundance analysis can be performed on cell type clusters, it’s not always possible or practical to use precisely labeled clusters, especially when we are interested in studying transitional states, such as during developmental processes, or when we expect only a subpopulation of a cell type to be affected by the condition of interest. Milo is a method to detect compositional changes occurring in smaller subpopulations of cells, defined as neighbourhoods over the k-nearest neighbor (KNN) graph of cell-cell similarities.
Build KNN graph¶
We can use omicverse functions to build a KNN graph. We set the dimensionality and value for k to use in subsequent steps.
Here the value of k indicates the smallest possible size of neighbourhood in which we will quantify differential abundance (i.e. with k=50 the smallest neighbourhood will have 50 cells). Depending on the number of samples, you might want to use a high value of k for neighbourhood analysis, to have sufficient power to estimate abundance fold-changes. Since here we have data from > 100 patients, we set k=150 to have on average more than one cell per donor in each neighbourhood.
ov.settings.cpu_gpu_mixed_init()
CPU-GPU mixed mode activated
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,
target_sum=50*1e4)
adata.raw = adata
adata = adata[:, adata.var.highly_variable_features]
ov.single.batch_correction(adata,batch_key='batch',
methods='harmony',n_pcs=50)
ov.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
use_rep='X_harmony')
ov.pp.umap(adata)
Begin robust gene identification
After filtration, 15215/15215 genes are kept. Among 15215 genes, 15215 genes are robust.
End of robust gene identification.
Begin size normalization: shiftlog and HVGs selection pearson
normalizing counts per cell. The following highly-expressed genes are not considered during normalization factor computation:
['Defa24', 'Fabp6', 'Gcg', 'Gip', 'Nts', 'Reg3b', 'Reg4', 'Sct', 'Spink4', 'Sst', 'Tff3', 'Zg16']
finished (0:00:00)
extracting highly variable genes
--> added
'highly_variable', boolean vector (adata.var)
'highly_variable_rank', float vector (adata.var)
'highly_variable_nbatches', int vector (adata.var)
'highly_variable_intersection', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
Time to analyze data in cpu: 7.434457540512085 seconds.
End of size normalization: shiftlog and HVGs selection pearson
...Begin using harmony to correct batch effect
🚀 Using GPU to calculate PCA...
📊 [GPU 0] ------------------------------ 5/12288 MiB (0.0%)
computing PCA🔍
with n_comps=50
finished✅ (0:00:01)
🖥️ Using Scanpy CPU to calculate neighbors...
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:02)
🔍 [2025-05-24 03:28:52] Running UMAP in 'cpu-gpu-mixed' mode...
🚀 Using torch GPU to calculate UMAP...
📊 [GPU 0] ------------------------------ 5/12288 MiB (0.0%)
computing UMAP🚀
finished ✅: added
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns) (0:00:05)
✅ UMAP completed successfully.
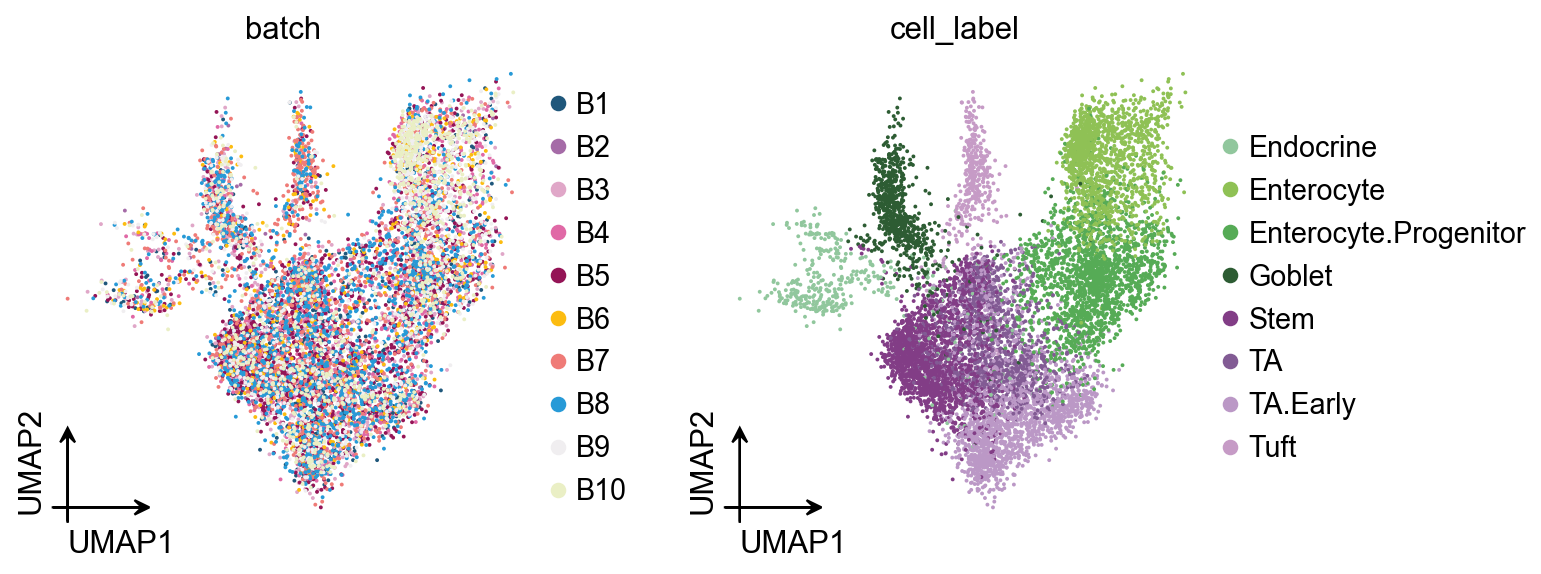
adata=ov.read('data/milo_test.h5ad')
ov.pl.umap(
adata,
color=['batch','cell_label'],
)
X_umap converted to UMAP to visualize and saved to adata.obsm['UMAP']
if you want to use X_umap, please set convert=False
Differential abundance testing with GLM¶
Similar to the scCODA approach, we use omicverse.single.DCT to perform differential cell–abundance analysis, except that here we set the method to milopy.
In releases after 1.7.9, we have removed the rpy2‐based edgeR dependency so that milo analyses can run entirely in a native Python environment. But we also keep the milo argument if user need the raw method
dct_obj=ov.single.DCT(
adata,
condition='condition',
ctrl_group='Control',
test_group='Salmonella',
cell_type_key='cell_label',
method='milopy',
sample_key='batch',
use_rep='X_harmony'
)
✅ Differential cell type abundance analysis initialized
📊 DCT analysis using milopy method
📊 Condition: condition, Control group: Control, Test group: Salmonella
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:21)
dct_obj.run()
✅ milopy DCT analysis completed
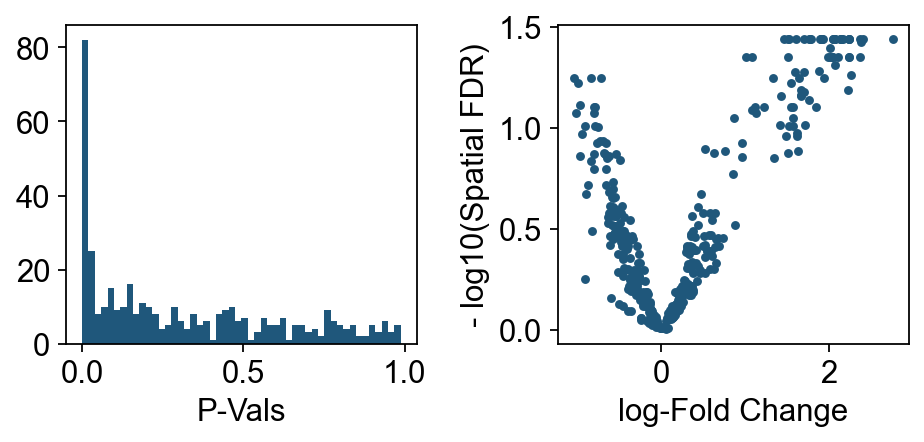
We can start inspecting the results of our DA analysis from a couple of standard diagnostic plots.
import matplotlib.pyplot as plt
old_figsize = plt.rcParams["figure.figsize"]
plt.rcParams["figure.figsize"] = [6, 3]
plt.subplot(1, 2, 1)
plt.hist(dct_obj.mdata["milo"].var.PValue, bins=50)
plt.xlabel("P-Vals")
plt.subplot(1, 2, 2)
plt.plot(dct_obj.mdata["milo"].var.logFC, -ov.np.log10(dct_obj.mdata["milo"].var.SpatialFDR), ".")
plt.xlabel("log-Fold Change")
plt.ylabel("- log10(Spatial FDR)")
plt.tight_layout()
plt.rcParams["figure.figsize"] = old_figsize
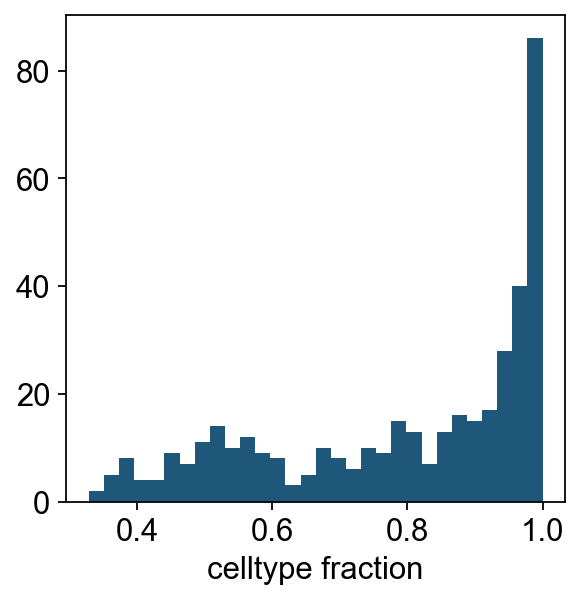
We can see that for the majority of neighbourhoods, almost all cells have the same cell type label. We can rename neighbourhoods where less than 60% of the cells have the top label as “Mixed”
plt.hist(dct_obj.mdata["milo"].var["nhood_annotation_frac"], bins=30)
plt.xlabel("celltype fraction")
Text(0.5, 0, 'celltype fraction')
res=dct_obj.get_results(mix_threshold=0.6)
res.head()
| index_cell | kth_distance | logFC | lfcSE | logCPM | stat | PValue | FDR | adj_pvalue | SpatialFDR | Nhood_size | nhood_annotation | nhood_annotation_frac | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | B1_AAACGCACTGTCCC_Control_Stem | 9.152347 | -0.583312 | 0.051568 | 11.412487 | 3.713398 | 0.080774 | 0.258990 | 0.258990 | 0.256133 | 399.0 | Mixed | 0.578947 |
| 1 | B1_AATAAGCTAGAGAT_Control_Enterocyte.Progenitor | 9.476911 | -0.809424 | 0.056194 | 11.429808 | 8.896907 | 0.012736 | 0.079455 | 0.079455 | 0.079067 | 408.0 | Mixed | 0.524510 |
| 2 | B1_ACGCTGCTCTCTTA_Control_Enterocyte.Progenitor | 9.004735 | 1.512198 | 0.017016 | 10.817280 | 12.689494 | 0.004606 | 0.045980 | 0.045980 | 0.044634 | 228.0 | Mixed | 0.557018 |
| 3 | B1_ACGGAACTGTTAGC_Control_Enterocyte.Progenitor | 9.966439 | 0.673496 | 0.032412 | 10.452936 | 2.316397 | 0.156838 | 0.362072 | 0.362072 | 0.350619 | 181.0 | Mixed | 0.563536 |
| 4 | B1_ACTTCTGATCGTTT_Control_TA.Early | 9.194638 | 0.202095 | 0.035912 | 11.222971 | 0.404716 | 0.537956 | 0.714916 | 0.714916 | 0.705203 | 339.0 | TA.Early | 0.820059 |
Visualization¶
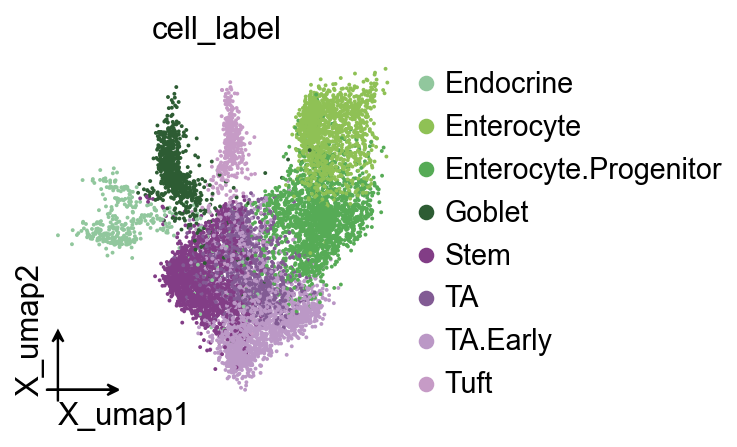
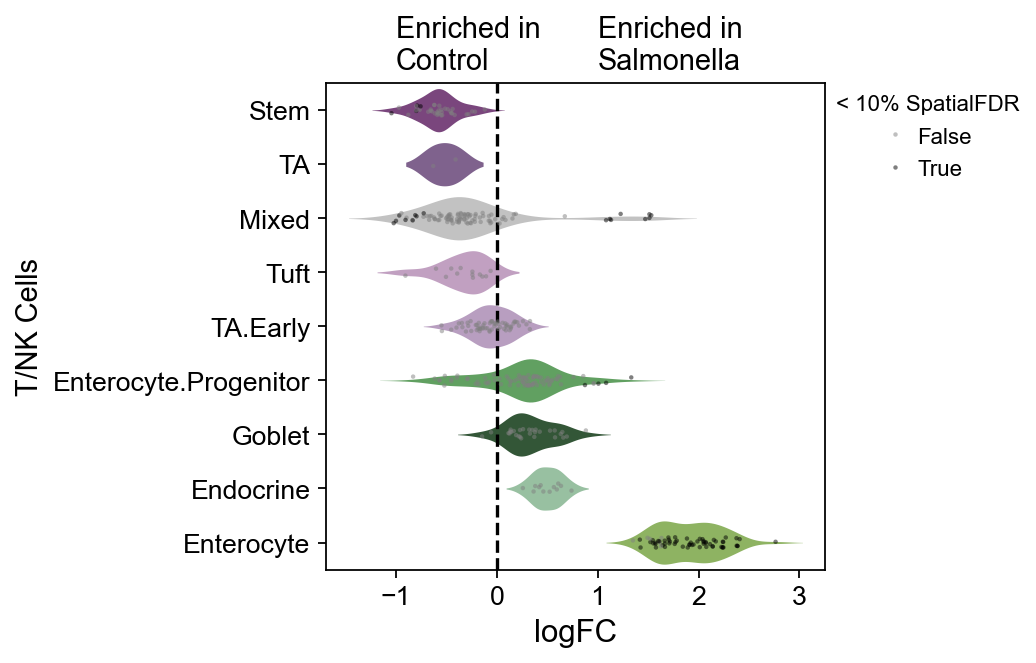
This is my favorite Milo plot. First, we create a color_dict to specify the cell colors we want to visualize.
color_dict=dict(zip(
adata.obs['cell_label'].cat.categories,
ov.pl.green_color[:4]+ov.pl.purple_color
))
color_dict['Mixed']='#c2c2c2'
fig, ax = plt.subplots(figsize=(3, 3))
ov.pl.embedding(
adata,
basis='X_umap',
color=['cell_label'],
palette=color_dict,
ax=ax,
#fig_size=(3,3)
)
#fig, ax = plt.subplots(figsize=(3, 4))
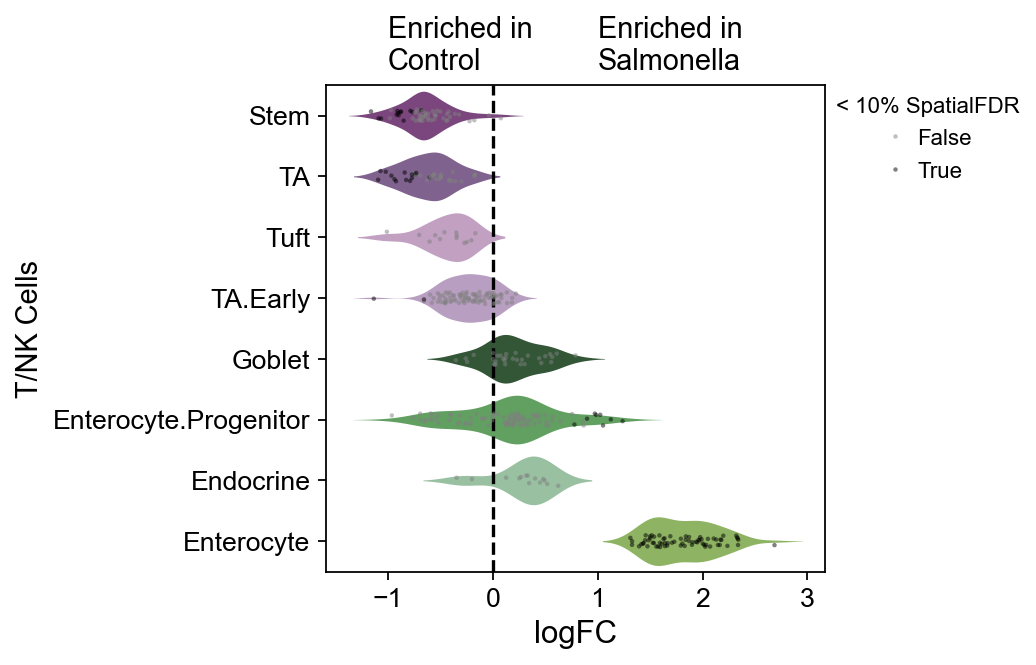
dct_obj.model.plot_da_beeswarm(
dct_obj.mdata,
alpha=0.1,
return_fig=True,
palette=color_dict,
)
ov.plt.xticks(fontsize=12)
ov.plt.yticks(fontsize=12)
ov.plt.ylabel('T/NK Cells',fontsize=13)
ov.plt.text(-1,-0.75,'Enriched in\nControl',fontsize=13)
ov.plt.text(1,-0.75,'Enriched in\nSalmonella',fontsize=13)
#fig
Text(1, -0.75, 'Enriched in\nSalmonella')
(Optional) DCT with milo¶
Tutorials could be found in https://pertpy.readthedocs.io/en/stable/tutorials/notebooks/milo.html
dct_obj_old=ov.single.DCT(
adata,
condition='condition',
ctrl_group='Control',
test_group='Salmonella',
cell_type_key='cell_label',
method='milo',
sample_key='batch',
use_rep='X_harmony'
)
✅ Differential cell type abundance analysis initialized
📊 DCT analysis using milo method
📊 Condition: condition, Control group: Control, Test group: Salmonella
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:07)
dct_obj_old.run()
✅ milo DCT analysis completed
#fig, ax = plt.subplots(figsize=(3, 4))
dct_obj_old.model.plot_da_beeswarm(
dct_obj_old.mdata,
alpha=0.1,
return_fig=True,
palette=color_dict,
)
ov.plt.xticks(fontsize=12)
ov.plt.yticks(fontsize=12)
ov.plt.ylabel('T/NK Cells',fontsize=13)
ov.plt.text(-1,-0.75,'Enriched in\nControl',fontsize=13)
ov.plt.text(1,-0.75,'Enriched in\nSalmonella',fontsize=13)
#fig
Text(1, -0.75, 'Enriched in\nSalmonella')