GeneFormer¶
Geneformer is a foundational transformer model pretrained on a large-scale corpus of single cell transcriptomes to enable context-aware predictions in settings with limited data in network biology.
Here, you can use omicverse.llm.SCLLMManager(model_type="geneformer") to call this model directly.
Cite: Theodoris, C. V., Xiao, L., Chopra, A., Chaffin, M. D., Al Sayed, Z. R., Hill, M. C., … & Ellinor, P. T. (2023). Transfer learning enables predictions in network biology. Nature, 618(7965), 616-624.
import scanpy as sc
import omicverse as ov
ov.plot_set(font_path='Arial')
# Enable auto-reload for development
%load_ext autoreload
%autoreload 2
🔬 Starting plot initialization...
Using already downloaded Arial font from: /tmp/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting CUDA devices…
✅ [GPU 0] NVIDIA H100 80GB HBM3
• Total memory: 79.1 GB
• Compute capability: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 1.7.6rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
Load example datasets¶
For this tutorial, we use three batches from the NeurIPS 2021 single-cell competition dataset, which provides an excellent test case for batch integration and cell type annotation.
adata1=ov.read('data/neurips2021_s1d3.h5ad')
adata1.obs['batch']='s1d3'
adata2=ov.read('data/neurips2021_s2d1.h5ad')
adata2.obs['batch']='s2d1'
adata3=ov.read('data/neurips2021_s3d7.h5ad')
adata3.obs['batch']='s3d7'
adata=sc.concat([adata1,adata2,adata3],merge='same')
adata
AnnData object with n_obs × n_vars = 27423 × 13953
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',
n_HVGs=3000,batch_key=None,target_sum=1e4)
adata
adata.raw = adata
adata = adata[:, adata.var.highly_variable_features]
adata
View of AnnData object with n_obs × n_vars = 27423 × 3000
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id', 'n_cells', 'percent_cells', 'robust', 'means', 'variances', 'residual_variances', 'highly_variable_rank', 'highly_variable_features'
uns: 'log1p', 'hvg', 'status', 'status_args', 'REFERENCE_MANU'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
Download pre-trained model and dictionaries¶
The Geneformer model requires several components:
Model weights: Pre-trained transformer parameters (~420MB)
Gene dictionaries: Mapping between genes and tokens
Gene median values: For rank-based encoding
Ensembl mappings: Gene symbol to ID conversions
Download from HuggingFace: https://huggingface.co/ctheodoris/Geneformer/tree/main/Geneformer-V2-104M
#!/usr/bin/env python3
import os
import requests
urls = [
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/Geneformer-V2-104M/model.safetensors?download=true",
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/Geneformer-V2-104M/config.json?download=true",
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/Geneformer-V2-104M/generation_config.json?download=true",
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/Geneformer-V2-104M/training_args.bin?download=true",
]
output_dir = "llm_model/models/geneformer/Geneformer-V2-104M"
os.makedirs(output_dir, exist_ok=True)
for url in urls:
filename = url.split('?')[0].split('/')[-1]
filepath = os.path.join(output_dir, filename)
print(f"Downloading {filename} ...")
resp = requests.get(url, stream=True)
resp.raise_for_status()
with open(filepath, "wb") as f:
for chunk in resp.iter_content(chunk_size=8192):
if chunk:
f.write(chunk)
print(f"Saved to {filepath}")
print("All files downloaded successfully.")
Downloading model.safetensors ...
Saved to llm_model/models/geneformer/Geneformer-V2-104M/model.safetensors
Downloading config.json ...
Saved to llm_model/models/geneformer/Geneformer-V2-104M/config.json
Downloading generation_config.json ...
Saved to llm_model/models/geneformer/Geneformer-V2-104M/generation_config.json
Downloading training_args.bin ...
Saved to llm_model/models/geneformer/Geneformer-V2-104M/training_args.bin
All files downloaded successfully.
#!/usr/bin/env python3
import os
import requests
urls = [
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/geneformer/ensembl_mapping_dict_gc104M.pkl?download=true",
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/geneformer/gene_median_dictionary_gc104M.pkl?download=true",
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/geneformer/gene_name_id_dict_gc104M.pkl?download=true",
"https://huggingface.co/ctheodoris/Geneformer/resolve/main/geneformer/token_dictionary_gc104M.pkl?download=true",
]
output_dir = "llm_model/models/geneformer"
os.makedirs(output_dir, exist_ok=True)
for url in urls:
filename = url.split('?')[0].split('/')[-1]
filepath = os.path.join(output_dir, filename)
print(f"Downloading {filename} ...")
resp = requests.get(url, stream=True)
resp.raise_for_status()
with open(filepath, "wb") as f:
for chunk in resp.iter_content(chunk_size=8192):
if chunk:
f.write(chunk)
print(f"Saved to {filepath}")
print("All files downloaded successfully.")
Downloading ensembl_mapping_dict_gc104M.pkl ...
Saved to llm_model/models/geneformer/ensembl_mapping_dict_gc104M.pkl
Downloading gene_median_dictionary_gc104M.pkl ...
Saved to llm_model/models/geneformer/gene_median_dictionary_gc104M.pkl
Downloading gene_name_id_dict_gc104M.pkl ...
Saved to llm_model/models/geneformer/gene_name_id_dict_gc104M.pkl
Downloading token_dictionary_gc104M.pkl ...
Saved to llm_model/models/geneformer/token_dictionary_gc104M.pkl
All files downloaded successfully.
Initialize Geneformer V2 model¶
Geneformer V2 is the latest version with improved performance and a larger parameter count (104M). The model architecture is based on BERT with modifications for single-cell data.
manager = ov.llm.SCLLMManager(
model_type='geneformer',
model_version='V2',
device='cuda',
)
#analysis/omic_test/llm_model/Geneformer/geneformer/ensembl_mapping_dict_gc104M.pkl
manager.model.load_model(
'llm_model/models/geneformer/Geneformer-V2-104M',
gene_median_file='llm_model/models/geneformer/gene_median_dictionary_gc104M.pkl',
token_dictionary_file='llm_model/models/geneformer/token_dictionary_gc104M.pkl',
gene_mapping_file='llm_model/models/geneformer/ensembl_mapping_dict_gc104M.pkl'
)
[Loaded] Geneformer model initialized (version: V2)
[Loading] Loading Geneformer model
Stored gene_median_file: llm_model/models/geneformer/gene_median_dictionary_gc104M.pkl
Stored token_dictionary_file: llm_model/models/geneformer/token_dictionary_gc104M.pkl
Stored gene_mapping_file: llm_model/models/geneformer/ensembl_mapping_dict_gc104M.pkl
[Loaded] Tokenizer initialized with external dictionary files
Gene median: gene_median_dictionary_gc104M.pkl
Token dictionary: token_dictionary_gc104M.pkl
[Loaded] Geneformer model loaded successfully
Version: V2
Zero-shot embedding generation¶
Generate embeddings using the pre-trained Geneformer model.
The resulting 768-dimensional embeddings capture cellular states based on gene expression patterns:
embeddings = manager.get_embeddings(adata,max_ncells=100000)
[🔬Cells] Data Summary:
Cells: 27,423
Genes: 3,000
Batches: 3
s3d7: 11,230 cells
s2d1: 10,258 cells
s1d3: 5,935 cells
[Embedding] Starting get_embeddings...
cells: 27,423
genes: 3,000
[Preprocessing] Preprocessing data for Geneformer...
normalizing counts per cell
finished (0:00:00)
[Loaded] Normalized total counts
[Preprocessing] Preprocessing completed: 27423 cells × 3000 genes
[Predicting] Extracting cell embeddings with Geneformer...
[Preprocessing] Converting data to Geneformer format
[Preprocessing] Preparing data for Geneformer tokenization
[Preprocessing] Adding ensembl_id column to adata.var
[Warning] Using gene symbols as ensembl_id (may cause filtering)
[ℹ️Info] Geneformer works best with Ensembl gene IDs
[ℹ️Info] Gene mapping analysis:
[Preprocessing] Proactive gene symbol mapping...
[Loaded] Successfully mapped 2767 genes to Ensembl IDs
[Warning] Adding n_counts column to adata.obs...
✓ Added n_counts: mean=9915.1, std=42.4
[Preprocessing] Adding cell_barcode column to preserve cell identity...
✓ Added cell_barcode column with 27423 barcodes
[Preprocessing] Tokenizing data for Geneformer
[Preprocessing] Attempting real Geneformer tokenization...
/tmp/tmpu4y82au8/input/temp_data.h5ad has no column attribute 'filter_pass'; tokenizing all cells.
[Loaded] Tokenized 27423 cells
Creating dataset.
[Training] Extracting embeddings...
[Loaded] Using all 27423 cells (preserving order)
[Loaded] Extracted embeddings from EmbExtractor: (27423, 768)
[✅Complete] get_embeddings completed successfully!
[✅Complete] Results summary:
embedding_shape: (27423, 768)
embedding_dim: 768
#adata.obsm['X_geneformer'] = df.loc[adata.obs.index,[f'emb_{i}' for i in range(0,768)]]
adata.obsm['X_geneformer'] = embeddings
sc.pp.neighbors(adata, use_rep='X_geneformer')
sc.tl.umap(adata)
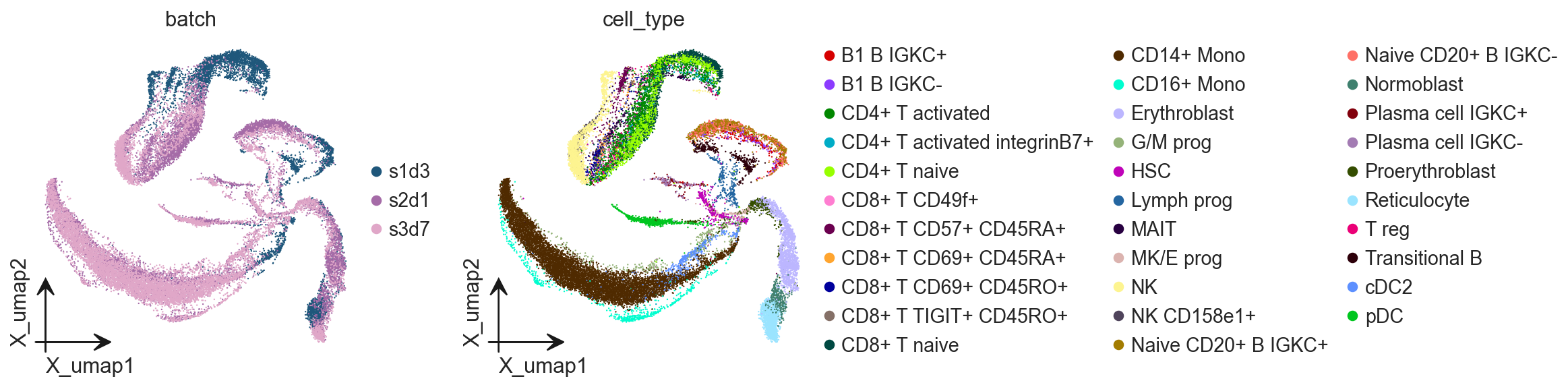
ov.pl.embedding(
adata,
basis='X_umap',
color=['batch', 'cell_type']
)
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:22)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns) (0:00:19)
Fine-tuning for cell type classification¶
Fine-tune Geneformer on a reference dataset to adapt it for specific cell type recognition. The fine-tuning process updates the classification head while keeping most transformer layers frozen to prevent overfitting.
reference_adata=adata[adata.obs['batch']=='s1d3']
reference_adata
View of AnnData object with n_obs × n_vars = 5935 × 3000
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id', 'n_cells', 'percent_cells', 'robust', 'means', 'variances', 'residual_variances', 'highly_variable_rank', 'highly_variable_features'
uns: 'log1p', 'hvg', 'status', 'status_args', 'REFERENCE_MANU'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
reference_adata.obs['celltype']=reference_adata.obs['cell_type'].copy()
fine_tune_results = manager.model.fine_tune(
train_adata=reference_adata,
epochs=10, #
batch_size=32, #
lr=1e-4, #
)
🔧 Fine-tuning Geneformer for annotation task...
Cell types detected: ['CD14+ Mono', 'CD8+ T naive', 'NK', 'T reg', 'CD8+ T CD57+ CD45RA+', 'Transitional B', 'Lymph prog', 'Naive CD20+ B IGKC+', 'Normoblast', 'Reticulocyte', 'CD4+ T naive', 'CD4+ T activated', 'CD8+ T TIGIT+ CD45RO+', 'B1 B IGKC+', 'CD4+ T activated integrinB7+', 'Erythroblast', 'MAIT', 'B1 B IGKC-', 'CD8+ T CD49f+', 'Naive CD20+ B IGKC-', 'G/M prog', 'Proerythroblast', 'HSC', 'CD16+ Mono', 'cDC2', 'CD8+ T CD69+ CD45RO+', 'pDC', 'CD8+ T CD69+ CD45RA+', 'Plasma cell IGKC+', 'MK/E prog']
[Preprocessing] Creating tokenized dataset
[Preprocessing] Preparing data for Geneformer tokenization
[Preprocessing] Adding ensembl_id column to adata.var
[Warning] Using gene symbols as ensembl_id (may cause filtering)
[ℹ️Info] Geneformer works best with Ensembl gene IDs
[ℹ️Info] Gene mapping analysis:
[Preprocessing] Proactive gene symbol mapping...
[Loaded] Successfully mapped 2767 genes to Ensembl IDs
[Warning] Adding n_counts column to adata.obs...
✓ Added n_counts: mean=1028.9, std=375.6
[Preprocessing] Adding cell_barcode column to preserve cell identity...
✓ Added cell_barcode column with 5935 barcodes
[Preprocessing] Tokenizing data for Geneformer
[Preprocessing] Attempting real Geneformer tokenization...
/tmp/tmpgf8hd0la/input/temp_data.h5ad has no column attribute 'filter_pass'; tokenizing all cells.
[Loaded] Tokenized 5935 cells
Creating dataset.
[Loaded] Tokenized 5935 cells
[Preprocessing] Adding labels and splitting dataset...
Using cell_barcode mapping for labels...
Train set: 5342 cells
Eval set: 593 cells
[Loaded] Model initialized with 30 classes
[Training] Starting training...
[✅Complete] Fine-tuning completed
[Loaded] Extracted BERT base model from classification model
| Epoch | Training Loss | Validation Loss | Accuracy | F1 |
|---|---|---|---|---|
| 1 | 0.830700 | 0.659676 | 0.833052 | 0.422297 |
| 2 | 0.410600 | 0.900503 | 0.726813 | 0.485091 |
| 3 | 0.682200 | 0.501471 | 0.871838 | 0.618438 |
| 4 | 0.309900 | 0.338666 | 0.900506 | 0.678833 |
| 5 | 0.523200 | 0.353199 | 0.895447 | 0.694306 |
| 6 | 0.209100 | 0.304860 | 0.913997 | 0.697800 |
| 7 | 0.137500 | 0.393189 | 0.885329 | 0.688280 |
| 8 | 0.100900 | 0.409528 | 0.905565 | 0.722270 |
| 9 | 0.132900 | 0.380685 | 0.885329 | 0.720899 |
| 10 | 0.083000 | 0.373825 | 0.903879 | 0.756619 |
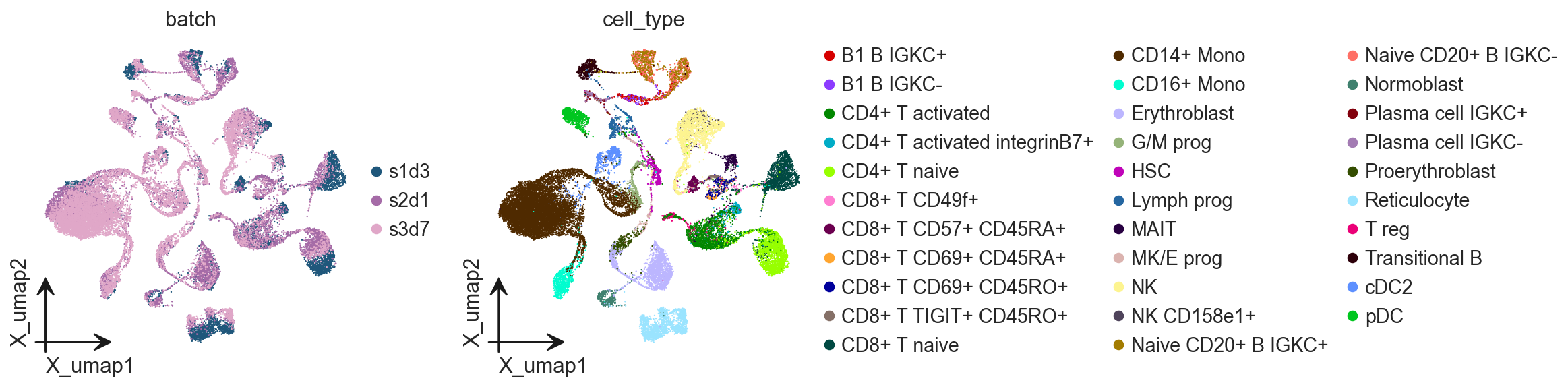
Batch integration with fine-tuned model¶
After fine-tuning, we perform batch integration to remove technical variations while preserving biological differences. This critical step ensures that cells from different batches can be properly compared and analyzed together.
zero_shot_results = manager.model.integrate(
adata,
batch_key="batch",
correction_method="mnn",
max_ncells=100000
)
adata.obsm['X_geneformer_fine'] = zero_shot_results['embeddings']
[Preprocessing] Performing batch integration with Geneformer embeddings...
[🔬Cells] Data Summary:
Cells: 27,423
Genes: 3,000
Batches: 3
s3d7: 11,230 cells
s2d1: 10,258 cells
s1d3: 5,935 cells
[Embedding] Starting get_embeddings...
cells: 27,423
genes: 3,000
[Preprocessing] Preprocessing data for Geneformer...
normalizing counts per cell
finished (0:00:00)
[Loaded] Normalized total counts
[Preprocessing] Preprocessing completed: 27423 cells × 3000 genes
[Predicting] Extracting cell embeddings with Geneformer...
[Preprocessing] Converting data to Geneformer format
[Preprocessing] Preparing data for Geneformer tokenization
[Preprocessing] Adding ensembl_id column to adata.var
[Warning] Using gene symbols as ensembl_id (may cause filtering)
[ℹ️Info] Geneformer works best with Ensembl gene IDs
[ℹ️Info] Gene mapping analysis:
[Preprocessing] Proactive gene symbol mapping...
[Loaded] Successfully mapped 2767 genes to Ensembl IDs
[Warning] Adding n_counts column to adata.obs...
✓ Added n_counts: mean=9915.1, std=42.4
[Preprocessing] Adding cell_barcode column to preserve cell identity...
✓ Added cell_barcode column with 27423 barcodes
[Preprocessing] Tokenizing data for Geneformer
[Preprocessing] Attempting real Geneformer tokenization...
/tmp/tmp6z_5sigv/input/temp_data.h5ad has no column attribute 'filter_pass'; tokenizing all cells.
[Loaded] Tokenized 27423 cells
Creating dataset.
[Training] Extracting embeddings...
[Loaded] Using all 27423 cells (preserving order)
[Loaded] Extracted embeddings from EmbExtractor: (27423, 768)
[✅Complete] get_embeddings completed successfully!
[✅Complete] Results summary:
embedding_shape: (27423, 768)
embedding_dim: 768
Found 3 batches with distribution: [ 5935 10258 11230]
Using raw Geneformer embeddings
[Loaded] Integration completed using mnn method
sc.pp.neighbors(adata, use_rep='X_geneformer_fine')
sc.tl.umap(adata)
ov.pl.embedding(
adata,
basis='X_umap',
color=['batch', 'cell_type']
)
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:04)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns) (0:00:19)
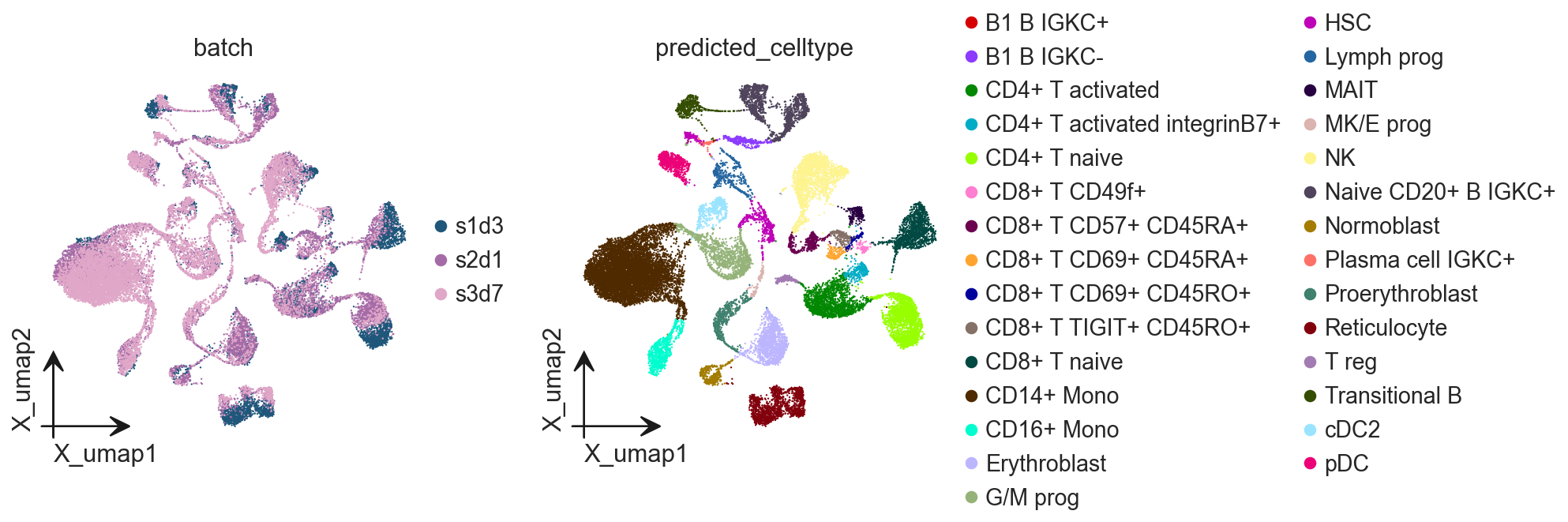
Cell type annotation with fine-tuned model¶
The fine-tuned GeneFormer model can now predict cell types for all cells in the dataset, including those from batches not used in training. This demonstrates the model’s ability to generalize learned patterns to new data while leveraging the improved discrimination capability gained through fine-tuning.
results_anno = manager.model.predict(
adata,
task="annotation",
max_ncells=100000
)
[Preprocessing] Preprocessing data for Geneformer...
normalizing counts per cell
finished (0:00:00)
[Loaded] Normalized total counts
[Preprocessing] Preprocessing completed: 27423 cells × 3000 genes
[Predicting] Predicting cell types with Geneformer...
[Preprocessing] Preparing data for prediction
[Preprocessing] Preparing data for Geneformer tokenization
[Preprocessing] Adding ensembl_id column to adata.var
[Warning] Using gene symbols as ensembl_id (may cause filtering)
[ℹ️Info] Geneformer works best with Ensembl gene IDs
[ℹ️Info] Gene mapping analysis:
[Preprocessing] Proactive gene symbol mapping...
[Loaded] Successfully mapped 2767 genes to Ensembl IDs
[Warning] Adding n_counts column to adata.obs...
✓ Added n_counts: mean=9915.1, std=42.4
[Preprocessing] Adding cell_barcode column to preserve cell identity...
✓ Added cell_barcode column with 27423 barcodes
[Preprocessing] Tokenizing data for Geneformer
[Preprocessing] Attempting real Geneformer tokenization...
/tmp/tmpm7wuqxu6/input/temp_data.h5ad has no column attribute 'filter_pass'; tokenizing all cells.
[Loaded] Tokenized 27423 cells
Creating dataset.
[Loaded] Tokenized 27423 cells for prediction
[Training] Running cell type prediction...
[Loaded] Predicted 27423 cells
adata.obs['predicted_celltype'] = results_anno['predicted_celltypes']
adata.obs['predicted_celltype_id'] = results_anno['predictions']
ov.pl.embedding(
adata,
basis='X_umap',
color=['batch', 'predicted_celltype']
)