Different Expression Analysis¶
An important task of bulk rna-seq analysis is the different expression , which we can perform with omicverse. For different expression analysis, ov change the gene_id to gene_name of matrix first. When our dataset existed the batch effect, we can use the SizeFactors of DEseq2 to normalize it, and use t-test of wilcoxon to calculate the p-value of genes. Here we demonstrate this pipeline with a matrix from featureCounts. The same pipeline would generally be used to analyze any collection of RNA-seq tasks.
Colab_Reproducibility:https://colab.research.google.com/drive/1q5lDfJepbtvNtc1TKz-h4wGUifTZ3i0_?usp=sharing
import omicverse as ov
import scanpy as sc
import matplotlib.pyplot as plt
ov.plot_set()
Warning: Could not read dependencies from pyproject.toml: [Errno 2] No such file or directory: '/home/groups/xiaojie/steorra/env/omicverse/lib/python3.10/site-packages/omicverse/pyproject.toml'
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
Version: 1.6.11, Tutorials: https://omicverse.readthedocs.io/
Geneset Download¶
When we need to convert a gene id, we need to prepare a mapping pair file. Here we have pre-processed 6 genome gtf files and generated mapping pairs including T2T-CHM13, GRCh38, GRCh37, GRCm39, danRer7, and danRer11. If you need to convert other id_mapping, you can generate your own mapping using gtf Place the files in the genesets directory.
ov.utils.download_geneid_annotation_pair()
......Geneid Annotation Pair download start: pair_GRCm39
......Loading dataset from genesets/pair_GRCm39.tsv
......Geneid Annotation Pair download start: pair_T2TCHM13
......Loading dataset from genesets/pair_T2TCHM13.tsv
......Geneid Annotation Pair download start: pair_GRCh38
......Loading dataset from genesets/pair_GRCh38.tsv
......Geneid Annotation Pair download start: pair_GRCh37
......Loading dataset from genesets/pair_GRCh37.tsv
......Geneid Annotation Pair download start: pair_danRer11
......Loading dataset from genesets/pair_danRer11.tsv
......Geneid Annotation Pair download start: pair_danRer7
......Loading dataset from genesets/pair_danRer7.tsv
......Geneid Annotation Pair download finished!
Note that this dataset has not been processed in any way and is only exported by featureCounts, and Sequence alignment was performed from the genome file of CRCm39
sample data can be download from: https://raw.githubusercontent.com/Starlitnightly/omicverse/master/sample/counts.txt
data=ov.pd.read_csv('https://raw.githubusercontent.com/Starlitnightly/omicverse/master/sample/counts.txt',index_col=0,sep='\t',header=1)
#data=ov.read('data/counts.txt',index_col=0,header=1)
#replace the columns `.bam` to ``
data.columns=[i.split('/')[-1].replace('.bam','') for i in data.columns]
data.head()
| 1--1 | 1--2 | 2--1 | 2--2 | 3--1 | 3--2 | 4--1 | 4--2 | 4-3 | 4-4 | Blank-1 | Blank-2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Geneid | ||||||||||||
| ENSMUSG00000102628 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 9 |
| ENSMUSG00000100595 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ENSMUSG00000097426 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| ENSMUSG00000104478 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ENSMUSG00000104385 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
ID mapping¶
We performed the gene_id mapping by the mapping pair file GRCm39 downloaded before.
data=ov.bulk.Matrix_ID_mapping(data,'genesets/pair_GRCm39.tsv')
data.head()
| 1--1 | 1--2 | 2--1 | 2--2 | 3--1 | 3--2 | 4--1 | 4--2 | 4-3 | 4-4 | Blank-1 | Blank-2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gm37732 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ENSMUSG00002075679 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mir485 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gm45619 | 6 | 1 | 6 | 3 | 3 | 9 | 4 | 2 | 22 | 3 | 0 | 17 |
| Gm25097 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Different expression analysis with ov¶
We can do differential expression analysis very simply by ov, simply by providing an expression matrix. To run DEG, we simply need to:
Read the raw count by featureCount or any other qualify methods.
Create an ov DEseq object.
dds=ov.bulk.pyDEG(data)
We notes that the gene_name mapping before exist some duplicates, we will process the duplicate indexes to retain only the highest expressed genes
dds.drop_duplicates_index()
print('... drop_duplicates_index success')
... drop_duplicates_index success
We also need to remove the batch effect of the expression matrix, estimateSizeFactors of DEseq2 to be used to normalize our matrix
dds.normalize()
print('... estimateSizeFactors and normalize success')
... estimateSizeFactors and normalize success
Now we can calculate the different expression gene from matrix, we need to input the treatment and control groups
ttest¶
treatment_groups=['4-3','4-4']
control_groups=['1--1','1--2']
result_ttest=dds.deg_analysis(treatment_groups,control_groups,method='ttest')
result_ttest.sort_values('qvalue').head()
⚙️ You are using ttest method for differential expression analysis.
⏰ Start to calculate qvalue...
✅ Differential expression analysis completed.
| pvalue | qvalue | FoldChange | MaxBaseMean | BaseMean | log2(BaseMean) | log2FC | abs(log2FC) | size | -log(pvalue) | -log(qvalue) | sig | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_id | ||||||||||||
| Uqcrh | 0.000005 | 0.000009 | 1.340669 | 2847.780642 | 2485.934617 | 11.279573 | 0.422953 | 0.422953 | 0.134067 | 5.341574 | 5.050298 | sig |
| Tdrd7 | 0.000016 | 0.000031 | 0.819932 | 570.613422 | 519.217650 | 9.020196 | -0.286423 | 0.286423 | 0.081993 | 4.796963 | 4.505703 | sig |
| Glo1-ps | 0.000016 | 0.000032 | 0.538839 | 1718.087949 | 1321.875745 | 10.368371 | -0.892074 | 0.892074 | 0.053884 | 4.792256 | 4.501011 | sig |
| Kmt2a | 0.000023 | 0.000045 | 1.203046 | 1337.209316 | 1224.344556 | 10.257794 | 0.266692 | 0.266692 | 0.120305 | 4.638583 | 4.347354 | sig |
| 5730522E02Rik | 0.000028 | 0.000054 | 34.728018 | 7.971628 | 3.985814 | 1.994874 | 5.118028 | 5.118028 | 3.472802 | 4.560393 | 4.270316 | sig |
(optional) edgeR with python¶
This module is a partial port in Python of the R Bioconductor edgeR package.
result_edgepy=dds.deg_analysis(treatment_groups,control_groups,method='edgepy')
result_edgepy.sort_values('qvalue').head()
⚙️ You are using edgepy method for differential expression analysis.
⏰ Start to create DGEList...
⏰ Start to calculate qvalue...
✅ Differential expression analysis completed.
| pvalue | qvalue | FoldChange | MaxBaseMean | BaseMean | log2(BaseMean) | log2FC | abs(log2FC) | size | -log(pvalue) | -log(qvalue) | sig | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_id | ||||||||||||
| Krtap9-5 | 2.904895e-46 | 1.583284e-41 | 0.000473 | 1547.775572 | 774.135593 | 9.596442 | -11.046357 | 11.046357 | 1.104636 | 45.536870 | 40.800441 | sig |
| Krtap31-2 | 2.824260e-36 | 7.696673e-32 | 0.005276 | 2119.945065 | 1065.447800 | 10.057244 | -7.566236 | 7.566236 | 0.756624 | 35.549095 | 31.113697 | sig |
| Krtap19-4 | 7.394568e-34 | 1.343445e-29 | 0.007051 | 1801.648929 | 907.058833 | 9.825052 | -7.147957 | 7.147957 | 0.714796 | 33.131087 | 28.871780 | sig |
| Gm4553 | 2.906425e-33 | 3.960294e-29 | 0.007580 | 1747.474564 | 880.242980 | 9.781758 | -7.043576 | 7.043576 | 0.704358 | 32.536641 | 28.402273 | sig |
| Gm10153 | 1.665008e-29 | 1.512493e-25 | 0.000884 | 267.271967 | 133.635984 | 7.062165 | -10.144441 | 10.144441 | 1.014444 | 28.778584 | 24.820307 | sig |
(optional) limma¶
This module is a partial port in Python of the R Bioconductor limma package.
result_limma=dds.deg_analysis(treatment_groups,control_groups,method='limma')
result_limma.sort_values('qvalue').head()
⚙️ You are using limma method for differential expression analysis.
⏰ Start to create DGEList...
⏰ Start to adjust pvalue...
✅ Differential expression analysis completed.
| pvalue | qvalue | FoldChange | MaxBaseMean | BaseMean | log2(BaseMean) | log2FC | abs(log2FC) | size | sig | -log(pvalue) | -log(qvalue) | F | t | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_id | ||||||||||||||
| Gm5853 | 0.000011 | 0.007723 | 5.216002 | 0.996454 | 0.498227 | -1.005126 | 2.382945 | 2.382945 | 0.238294 | sig | 4.951007 | 2.112206 | 31871.910979 | 178.527060 |
| Gm14409 | 0.000011 | 0.007723 | 5.216002 | 0.996454 | 0.498227 | -1.005126 | 2.382945 | 2.382945 | 0.238294 | sig | 4.951007 | 2.112206 | 31871.910979 | 178.527060 |
| Tdrd7 | 0.000005 | 0.007723 | 0.819932 | 570.613422 | 519.217650 | 9.020196 | -0.286423 | 0.286423 | 0.028642 | sig | 5.330687 | 2.112206 | 69832.580356 | -264.258548 |
| Myl10 | 0.000009 | 0.007723 | 13.648007 | 2.989361 | 1.494680 | 0.579837 | 3.770618 | 3.770618 | 0.377062 | sig | 5.052672 | 2.112206 | 39320.908935 | 198.295005 |
| 5730522E02Rik | 0.000009 | 0.007723 | 34.728018 | 7.971628 | 3.985814 | 1.994874 | 5.118028 | 5.118028 | 0.511803 | sig | 5.064980 | 2.112206 | 40333.534198 | 200.832105 |
One important thing is that we do not filter out low expression genes when processing DEGs, and in future versions I will consider building in the corresponding processing.
print(dds.result.shape)
dds.result=dds.result.loc[dds.result['log2(BaseMean)']>1]
print(dds.result.shape)
(54504, 14)
(21279, 14)
We also need to set the threshold of Foldchange, we prepare a method named foldchange_set to finish. This function automatically calculates the appropriate threshold based on the log2FC distribution, but you can also enter it manually.
# -1 means automatically calculates
dds.foldchange_set(fc_threshold=-1,
pval_threshold=0.05,
logp_max=6)
... Fold change threshold: 1.569942639522294
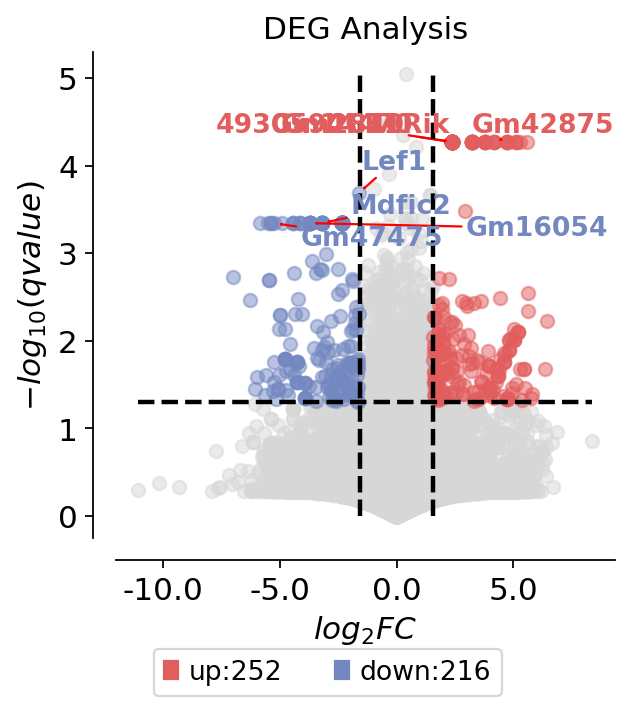
Visualize the DEG result and specific genes¶
To visualize the DEG result, we use plot_volcano to do it. This fuction can visualize the gene interested or high different expression genes. There are some parameters you need to input:
title: The title of volcano
figsize: The size of figure
plot_genes: The genes you interested
plot_genes_num: If you don’t have interested genes, you can auto plot it.
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
plot_genes_num=8,plot_genes_fontsize=12,)
<AxesSubplot: title={'center': 'DEG Analysis'}, xlabel='$log_{2}FC$', ylabel='$-log_{10}(qvalue)$'>
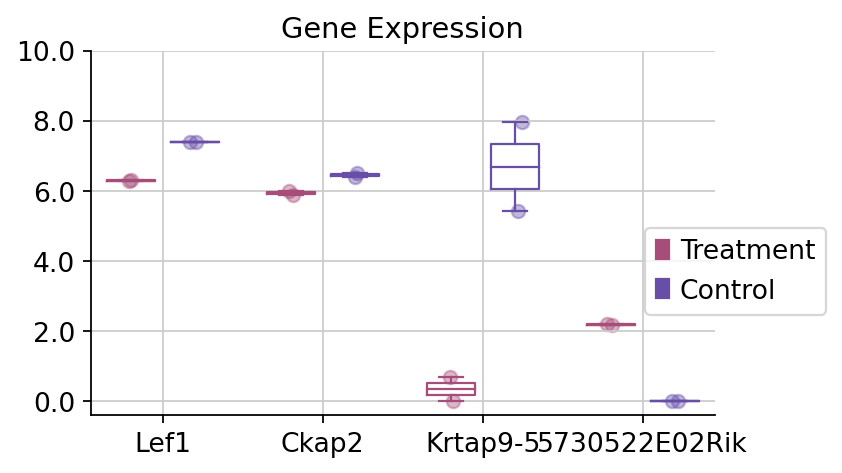
To visualize the specific genes, we only need to use the dds.plot_boxplot function to finish it.
dds.plot_boxplot(genes=['Ckap2','Lef1','Krtap9-5','5730522E02Rik'],treatment_groups=treatment_groups,
control_groups=control_groups,figsize=(5,3),fontsize=12,
legend_bbox=(1.2,0.55))
(<Figure size 400x240 with 1 Axes>,
<AxesSubplot: title={'center': 'Gene Expression'}>)
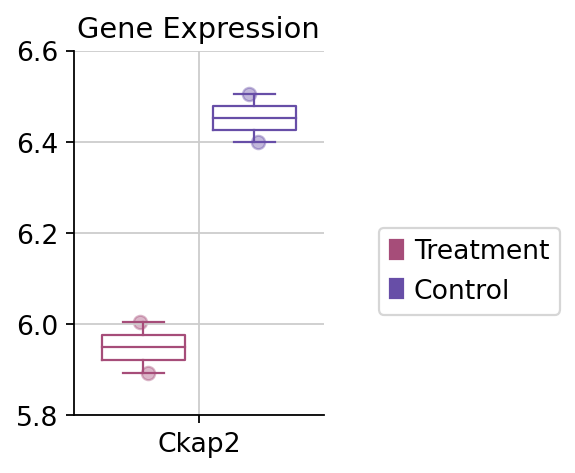
fig,ax=dds.plot_boxplot(genes=['Ckap2'],treatment_groups=treatment_groups,
control_groups=control_groups,figsize=(2,3),fontsize=12,
legend_bbox=(2,0.55))
#convert the ytickbels
ax.set_yticklabels([round(float(i.get_text()),2) for i in ax.get_yticklabels()])
[Text(0, 5.800000000000001, '5.8'),
Text(0, 6.000000000000001, '6.0'),
Text(0, 6.200000000000001, '6.2'),
Text(0, 6.4, '6.4'),
Text(0, 6.6000000000000005, '6.6')]
Pathway enrichment analysis by ov¶
Here we use the gseapy package, which included the GSEA analysis and Enrichment. We have optimised the output of the package and given some better looking graph drawing functions
Similarly, we need to download the pathway/genesets first. Five genesets we prepare previously, you can use ov.utils.download_pathway_database() to download automatically. Besides, you can download the pathway you interested from enrichr: https://maayanlab.cloud/Enrichr/#libraries
ov.utils.download_pathway_database()
......Pathway Geneset download start: GO_Biological_Process_2021
......Loading dataset from genesets/GO_Biological_Process_2021.txt
......Pathway Geneset download start: GO_Cellular_Component_2021
......Loading dataset from genesets/GO_Cellular_Component_2021.txt
......Pathway Geneset download start: GO_Molecular_Function_2021
......Loading dataset from genesets/GO_Molecular_Function_2021.txt
......Pathway Geneset download start: WikiPathway_2021_Human
......Loading dataset from genesets/WikiPathway_2021_Human.txt
......Pathway Geneset download start: WikiPathways_2019_Mouse
......Loading dataset from genesets/WikiPathways_2019_Mouse.txt
......Pathway Geneset download start: Reactome_2022
......Loading dataset from genesets/Reactome_2022.txt
......Pathway Geneset download finished!
pathway_dict=ov.utils.geneset_prepare('genesets/WikiPathways_2019_Mouse.txt',organism='Mouse')
Note that the pvalue_type we set to auto, this is because when the genesets we enrichment if too small, use the adjusted pvalue we can’t get the correct result. So you can set adjust or raw to get the significant geneset.
If you didn’t have internet, please set background to all genes expressed in rna-seq,like:
enr=ov.bulk.geneset_enrichment(gene_list=deg_genes,
pathways_dict=pathway_dict,
pvalue_type='auto',
background=dds.result.index.tolist(),
organism='mouse')
deg_genes=dds.result.loc[dds.result['sig']!='normal'].index.tolist()
enr=ov.bulk.geneset_enrichment(gene_list=deg_genes,
pathways_dict=pathway_dict,
pvalue_type='auto',
organism='mouse')
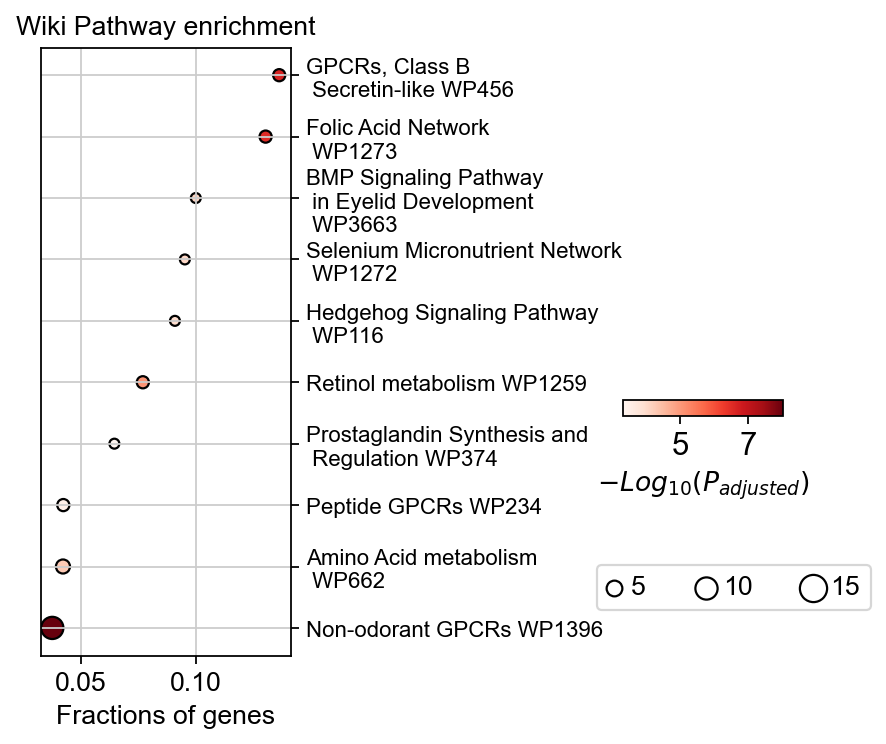
To visualize the enrichment, we use geneset_plot to finish it
ov.bulk.geneset_plot(enr,figsize=(2,5),fig_title='Wiki Pathway enrichment',
cax_loc=[2, 0.45, 0.5, 0.02],
bbox_to_anchor_used=(-0.25, -13),node_diameter=10,
custom_ticks=[5,7],text_knock=3,
cmap='Reds')
<AxesSubplot: title={'center': 'Wiki Pathway enrichment'}, xlabel='Fractions of genes'>
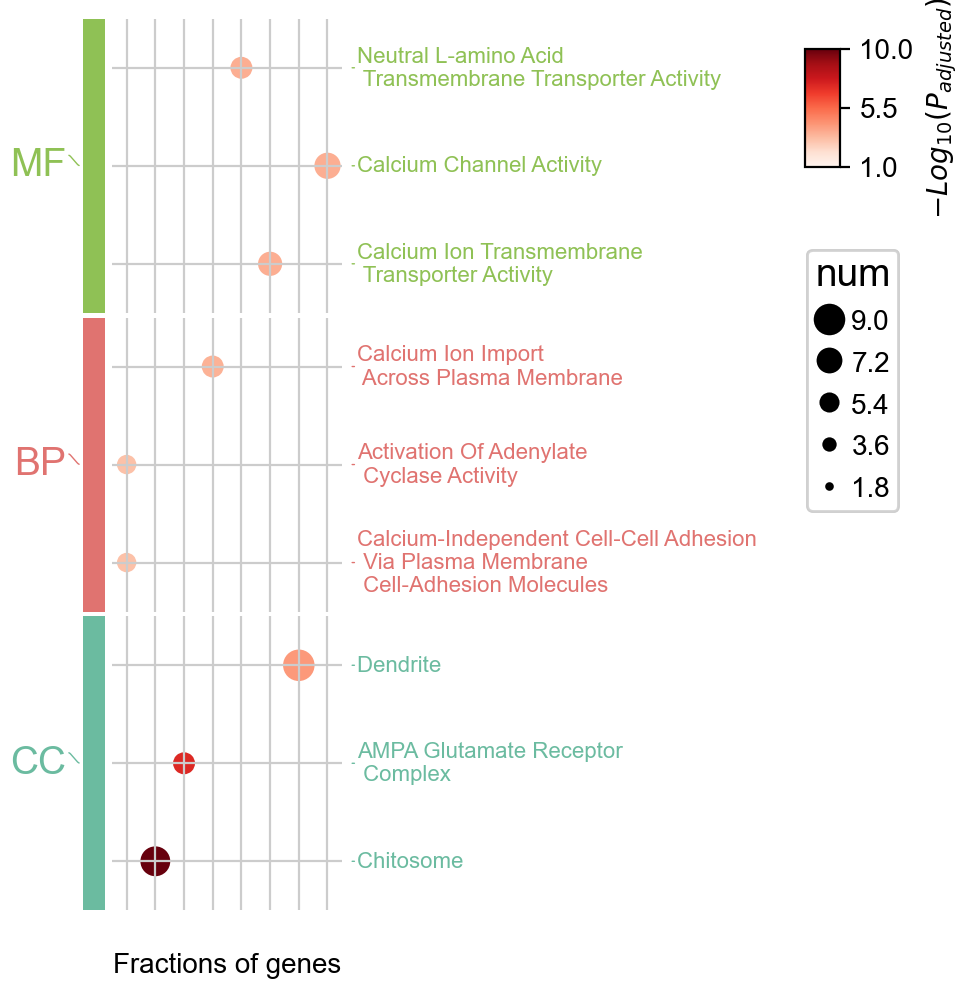
Multi pathway enrichment¶
In addition to pathway enrichment for a single database, OmicVerse supports enriching and visualizing multiple pathways at the same time, which is implemented using pyComplexHeatmap, and Citetation is welcome!
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Biological_Process_2023.txt',organism='Mouse')
enr_go_bp=ov.bulk.geneset_enrichment(gene_list=deg_genes,
pathways_dict=pathway_dict,
pvalue_type='auto',
organism='mouse')
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Molecular_Function_2023.txt',organism='Mouse')
enr_go_mf=ov.bulk.geneset_enrichment(gene_list=deg_genes,
pathways_dict=pathway_dict,
pvalue_type='auto',
organism='mouse')
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Cellular_Component_2023.txt',organism='Mouse')
enr_go_cc=ov.bulk.geneset_enrichment(gene_list=deg_genes,
pathways_dict=pathway_dict,
pvalue_type='auto',
organism='mouse')
enr_dict={'BP':enr_go_bp,
'MF':enr_go_mf,
'CC':enr_go_cc}
colors_dict={
'BP':ov.pl.red_color[1],
'MF':ov.pl.green_color[1],
'CC':ov.pl.blue_color[1],
}
ov.bulk.geneset_plot_multi(enr_dict,colors_dict,num=3,
figsize=(2,5),
text_knock=3,fontsize=8,
cmap='Reds'
)
Starting plotting..
Starting calculating row orders..
Reordering rows..
Starting calculating col orders..
Reordering cols..
Plotting matrix..
Inferred max_s (max size of scatter point) is: 106.70147475919528
Collecting legends..
Plotting legends..
Estimated legend width: 9.879166666666666 mm
<Axes: ylabel='$−Log_{10}(P_{adjusted})$'>