Batch correction in Bulk RNA-seq or microarray data¶
Variability in datasets are not only the product of biological processes: they are also the product of technical biases (Lander et al, 1999). ComBat is one of the most widely used tool for correcting those technical biases called batch effects.
pyComBat (Behdenna et al, 2020) is a new Python implementation of ComBat (Johnson et al, 2007), a software widely used for the adjustment of batch effects in microarray data. While the mathematical framework is strictly the same, pyComBat:
has similar results in terms of batch effects correction;
is as fast or faster than the R implementation of ComBat and;
offers new tools for the community to participate in its development.
Code: https://github.com/epigenelabs/pyComBat
Colab_Reproducibility:https://colab.research.google.com/drive/121bbIiI3j4pTZ3yA_5p8BRkRyGMMmNAq?usp=sharing
import anndata
import pandas as pd
import omicverse as ov
ov.ov_plot_set()
AnnData object with n_obs × n_vars = 17126 × 161
Loading dataset¶
This minimal usage example illustrates how to use pyComBat in a default setting, and shows some results on ovarian cancer data, freely available on NCBI’s Gene Expression Omnibus, namely:
GSE18520
GSE66957
GSE69428
The corresponding expression files are available on GitHub.
dataset_1 = pd.read_pickle("data/combat/GSE18520.pickle")
adata1=anndata.AnnData(dataset_1.T)
adata1.obs['batch']='1'
adata1
AnnData object with n_obs × n_vars = 63 × 21755
obs: 'batch'
dataset_2 = pd.read_pickle("data/combat/GSE66957.pickle")
adata2=anndata.AnnData(dataset_2.T)
adata2.obs['batch']='2'
adata2
AnnData object with n_obs × n_vars = 69 × 22115
obs: 'batch'
dataset_3 = pd.read_pickle("data/combat/GSE69428.pickle")
adata3=anndata.AnnData(dataset_3.T)
adata3.obs['batch']='3'
adata3
AnnData object with n_obs × n_vars = 29 × 21755
obs: 'batch'
We use the concat function to join the three datasets together and take the intersection for the same genes
adata=anndata.concat([adata1,adata2,adata3],merge='same')
adata
AnnData object with n_obs × n_vars = 161 × 17126
obs: 'batch'
Removing batch effect¶
ov.bulk.batch_correction(adata,batch_key='batch')
Found 3 batches.
Adjusting for 0 covariate(s) or covariate level(s).
Standardizing Data across genes.
Fitting L/S model and finding priors.
Finding parametric adjustments.
Adjusting the Data
Storing batch correction result in adata.layers['batch_correction']
Saving results¶
Raw datasets
raw_data=adata.to_df().T
raw_data.head()
| GSM461348 | GSM461349 | GSM461350 | GSM461351 | GSM461352 | GSM461353 | GSM461354 | GSM461355 | GSM461356 | GSM461357 | ... | GSM1701044 | GSM1701045 | GSM1701046 | GSM1701047 | GSM1701048 | GSM1701049 | GSM1701050 | GSM1701051 | GSM1701052 | GSM1701053 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_symbol | |||||||||||||||||||||
| A1BG | 4.140079 | 4.589471 | 4.526200 | 4.326366 | 4.141506 | 4.528423 | 4.419378 | 4.345215 | 4.184150 | 4.393646 | ... | 3.490229 | 4.542913 | 4.654638 | 4.199212 | 4.080964 | 4.114272 | 3.883770 | 4.103220 | 3.883770 | 3.487520 |
| A1BG-AS1 | 5.747137 | 6.130257 | 5.781449 | 5.914044 | 6.277715 | 5.668244 | 5.879830 | 6.013979 | 5.968187 | 6.017624 | ... | 4.005230 | 4.301880 | 4.509698 | 4.089223 | 4.129560 | 3.867568 | 4.094032 | 3.616044 | 4.307225 | 3.891060 |
| A1CF | 5.026369 | 5.120523 | 5.220462 | 4.828303 | 5.078094 | 5.204209 | 4.865024 | 5.119230 | 5.219517 | 4.706891 | ... | 4.225589 | 3.530307 | 3.215182 | 2.967515 | 3.012953 | 3.496765 | 3.117002 | 3.072093 | 2.570765 | 3.163533 |
| A2M | 7.892506 | 7.730116 | 7.796338 | 8.525167 | 7.545033 | 7.846979 | 7.638513 | 7.487679 | 7.533089 | 6.965395 | ... | 10.273206 | 4.061912 | 4.393332 | 4.716536 | 3.447348 | 3.134037 | 4.009413 | 3.953612 | 7.664853 | 3.548574 |
| A2ML1 | 3.966217 | 4.482255 | 3.964664 | 3.906967 | 3.952821 | 3.985276 | 3.997008 | 4.101457 | 4.015285 | 3.765736 | ... | 2.478731 | 4.132282 | 3.952693 | 2.527621 | 2.358378 | 2.414869 | 2.204600 | 2.295500 | 2.167646 | 2.216867 |
5 rows × 161 columns
Removing Batch datasets
removing_data=adata.to_df(layer='batch_correction').T
removing_data.head()
| GSM461348 | GSM461349 | GSM461350 | GSM461351 | GSM461352 | GSM461353 | GSM461354 | GSM461355 | GSM461356 | GSM461357 | ... | GSM1701044 | GSM1701045 | GSM1701046 | GSM1701047 | GSM1701048 | GSM1701049 | GSM1701050 | GSM1701051 | GSM1701052 | GSM1701053 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_symbol | |||||||||||||||||||||
| A1BG | 4.223549 | 4.846659 | 4.758930 | 4.481847 | 4.225527 | 4.762011 | 4.610814 | 4.507982 | 4.284656 | 4.575134 | ... | 4.237836 | 5.378695 | 5.499778 | 5.006204 | 4.878052 | 4.914150 | 4.664341 | 4.902172 | 4.664341 | 4.234900 |
| A1BG-AS1 | 5.730287 | 6.253722 | 5.777166 | 5.958322 | 6.455185 | 5.622501 | 5.911578 | 6.094858 | 6.032295 | 6.099838 | ... | 5.841898 | 5.990944 | 6.095359 | 5.884098 | 5.904365 | 5.772731 | 5.886515 | 5.646358 | 5.993630 | 5.784535 |
| A1CF | 3.922941 | 3.975597 | 4.031489 | 3.812171 | 3.951869 | 4.022399 | 3.832708 | 3.974874 | 4.030960 | 3.744271 | ... | 4.229097 | 3.822095 | 3.637628 | 3.492649 | 3.519248 | 3.802460 | 3.580155 | 3.553867 | 3.260401 | 3.607394 |
| A2M | 9.488789 | 9.219466 | 9.329295 | 10.538060 | 8.912504 | 9.413282 | 9.067542 | 8.817383 | 8.892696 | 7.951175 | ... | 11.137033 | 7.182184 | 7.393206 | 7.598996 | 6.790880 | 6.591389 | 7.148757 | 7.113228 | 9.476245 | 6.855333 |
| A2ML1 | 4.317770 | 5.553678 | 4.314051 | 4.175866 | 4.285686 | 4.363418 | 4.391514 | 4.641670 | 4.435287 | 3.837621 | ... | 3.807064 | 5.766146 | 5.553374 | 3.864987 | 3.664473 | 3.731402 | 3.482281 | 3.589976 | 3.438499 | 3.496814 |
5 rows × 161 columns
save
raw_data.to_csv('raw_data.csv')
removing_data.to_csv('removing_data.csv')
You can also save adata object
adata.write_h5ad('adata_batch.h5ad',compression='gzip')
#adata=ov.read('adata_batch.h5ad')
Compare the dataset before and after correction¶
We specify three different colours for three different datasets
color_dict={
'1':ov.utils.red_color[1],
'2':ov.utils.blue_color[1],
'3':ov.utils.green_color[1],
}
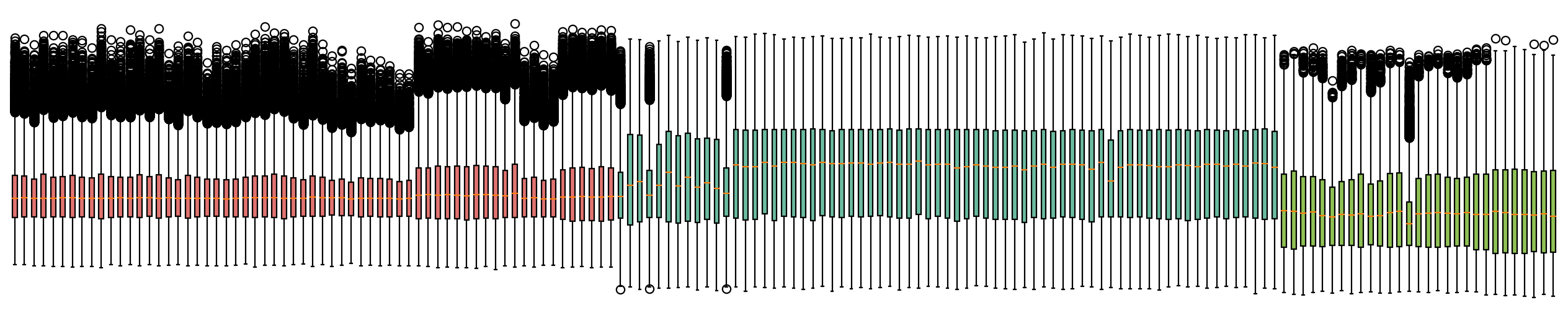
fig,ax=plt.subplots( figsize = (20,4))
bp=plt.boxplot(adata.to_df().T,patch_artist=True)
for i,batch in zip(range(adata.shape[0]),adata.obs['batch']):
bp['boxes'][i].set_facecolor(color_dict[batch])
ax.axis(False)
plt.show()
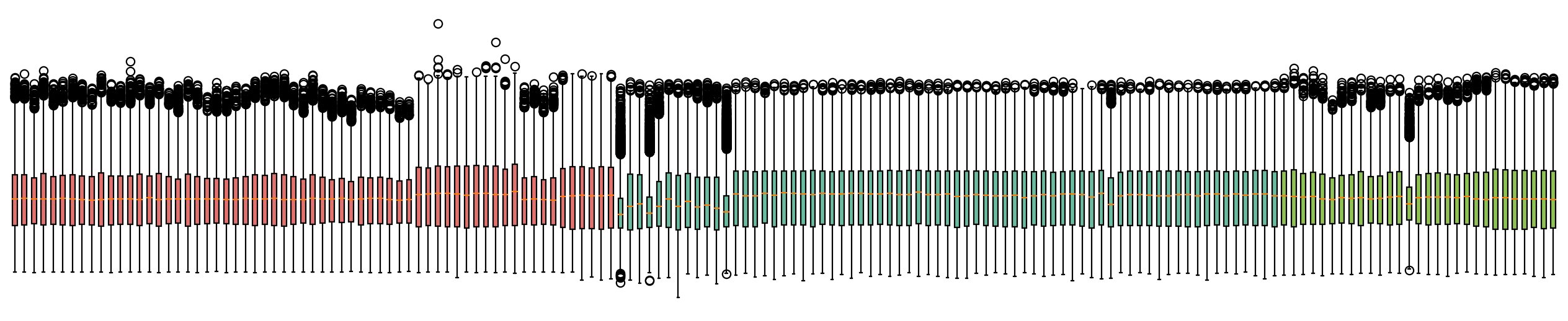
fig,ax=plt.subplots( figsize = (20,4))
bp=plt.boxplot(adata.to_df(layer='batch_correction').T,patch_artist=True)
for i,batch in zip(range(adata.shape[0]),adata.obs['batch']):
bp['boxes'][i].set_facecolor(color_dict[batch])
ax.axis(False)
plt.show()
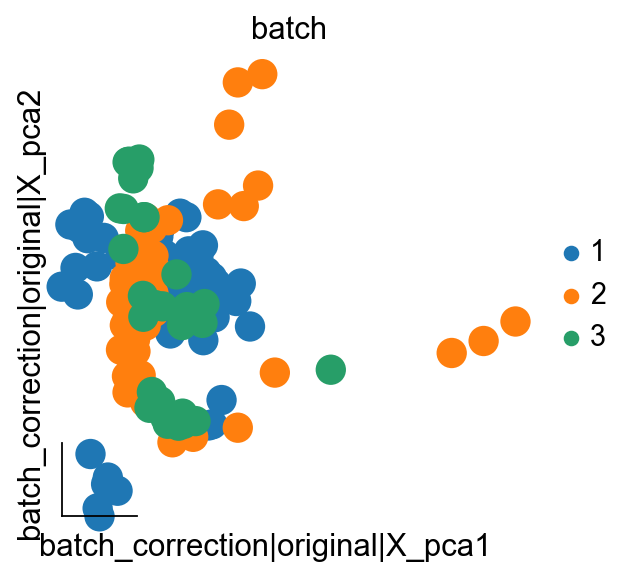
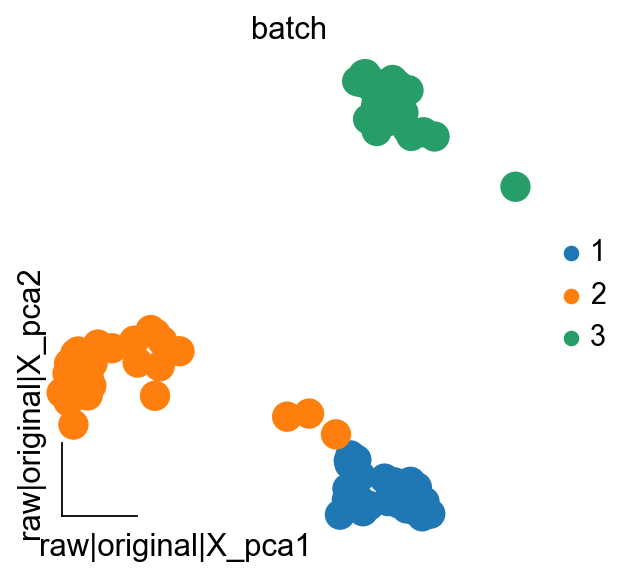
In addition to using boxplots to observe the effect of batch removal, we can also use PCA to observe the effect of batch removal
adata.layers['raw']=adata.X.copy()
We first calculate the PCA on the raw dataset
ov.pp.pca(adata,layer='raw',n_pcs=50)
adata
AnnData object with n_obs × n_vars = 161 × 17126
obs: 'batch'
uns: 'raw|original|pca_var_ratios', 'raw|original|cum_sum_eigenvalues'
obsm: 'raw|original|X_pca'
varm: 'raw|original|pca_loadings'
layers: 'batch_correction', 'raw', 'lognorm'
We then calculate the PCA on the batch_correction dataset
ov.pp.pca(adata,layer='batch_correction',n_pcs=50)
adata
AnnData object with n_obs × n_vars = 161 × 17126
obs: 'batch'
uns: 'raw|original|pca_var_ratios', 'raw|original|cum_sum_eigenvalues', 'batch_correction|original|pca_var_ratios', 'batch_correction|original|cum_sum_eigenvalues'
obsm: 'raw|original|X_pca', 'batch_correction|original|X_pca'
varm: 'raw|original|pca_loadings', 'batch_correction|original|pca_loadings'
layers: 'batch_correction', 'raw', 'lognorm'
ov.pl.embedding(adata,
basis='raw|original|X_pca',
color='batch',
frameon='small')
ov.pl.embedding(adata,
basis='batch_correction|original|X_pca',
color='batch',
frameon='small')