RNA-seq分析: 通路富集分析(GSEA)
在得到差异表达分析的结果后,我们往往还会对差异表达基因相关的功能感兴趣,在这里,我们介绍GSEA方法,这是一种比较好用的通路富集方法,可以根据基因表达的倍数情况,来分析基因相关的通路。
一般来说,做GSEA分析其实是可以不用做提取差异表达基因分析的结果的,直接用DESeq2的结果就可以了,但是我这里更关注差异表达基因相关的功能,所以就用了差异表达基因来做GSEA分析
1. 数据预处理
1.1 加载包
import gseapy as gp
1.2 提取数据
由于我们需要同时考虑log2FoldChange与p-value,所以我们这里用一个新的公式来计算基因倍数
metric=-sign(log2FoldChange)/-lg10(pvalue)
gseada=data1.loc[data1['sig']!='normal']
#倍数变化规则
gseada['fcsign']=-np.sign(gseada['log2FoldChange'])
gseada['logp']=-np.log10(gseada['pvalue'])
gseada['metric']=gseada['logp']/gseada['fcsign']
gseada.head()
得到结果如下
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | sig | log(padj) | fcsign | logp | metric | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID3 | 5442.355360 | -5.710849 | 0.145082 | -39.362892 | 0.000000e+00 | 0.000000e+00 | down | inf | 1.0 | inf | inf |
| TXNIP | 3147.115496 | 4.041551 | 0.103068 | 39.212444 | 0.000000e+00 | 0.000000e+00 | up | inf | -1.0 | inf | -inf |
| MMP1 | 4344.077177 | 4.476829 | 0.108971 | 41.082873 | 0.000000e+00 | 0.000000e+00 | up | inf | -1.0 | inf | -inf |
| ID1 | 5526.691818 | -5.280627 | 0.112286 | -47.028177 | 0.000000e+00 | 0.000000e+00 | down | inf | 1.0 | inf | inf |
| PTGIS | 4187.607799 | 3.441545 | 0.092530 | 37.193624 | 8.651991e-303 | 3.367009e-299 | up | 298.472756 | -1.0 | 302.062884 | -302.062884 |
1.3 数据排序
gseada=gseada.sort_values(by=['metric'],ascending=False)
gseada.head()
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | sig | log(padj) | fcsign | logp | metric | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID3 | 5442.355360 | -5.710849 | 0.145082 | -39.362892 | 0.000000e+00 | 0.000000e+00 | down | inf | 1.0 | inf | inf |
| ID1 | 5526.691818 | -5.280627 | 0.112286 | -47.028177 | 0.000000e+00 | 0.000000e+00 | down | inf | 1.0 | inf | inf |
| OXTR | 4105.199915 | -2.959533 | 0.100562 | -29.429803 | 2.283138e-190 | 2.468072e-187 | down | 186.607642 | 1.0 | 189.641468 | 189.641468 |
| SAMD11 | 1574.582498 | -6.502040 | 0.235542 | -27.604533 | 9.816869e-168 | 8.682574e-165 | down | 164.061352 | 1.0 | 167.008027 | 167.008027 |
1.4 rnk矩阵提取
我们发现数据中有inf,这对后续的分析有一定的影响,所以我们对inf进行赋值。
rnk=pd.DataFrame()
rnk['gene_name']=gseada.index
rnk['rnk']=gseada['metric'].values
k=1
total=0
for i in range(len(rnk)):
if rnk.loc[i,'rnk']==np.inf:
total+=1
#200跟274根据你的数据进行更改,保证inf比你数据最大的大,-inf比数据最小的小就好
for i in range(len(rnk)):
if rnk.loc[i,'rnk']==np.inf:
rnk.loc[i,'rnk']=200+(total-k)
k+=1
elif rnk.loc[i,'rnk']==-np.inf:
rnk.loc[i,'rnk']=-(274+k)
k+=1
#rnk=rnk.replace(np.inf,300)
#rnk=rnk.replace(-np.inf,-300)
rnk.head()
| gene_name | rnk | |
|---|---|---|
| 0 | ID3 | 201.000000 |
| 1 | ID1 | 200.000000 |
| 2 | OXTR | 189.641468 |
| 3 | SAMD11 | 167.008027 |
| 4 | ATOH8 | 146.165895 |
2. GSEA通路富集
2.1 基因index转大写
由于gseapy的包只能识别大写的基因名,所以我们需要先把所有的基因名转换成大写的。
for i in range(len(rnk)):
rnk.loc[i,'gene_name']=rnk.loc[i,'gene_name'].upper()
rnk.head()
2.2 GSEA通路富集
这里简单介绍以下函数的参数
rnk:输入排序矩阵
gene_sets:需要富集到的通路数据集
processes:并行使用的线程数
permutation_num:检验的速度
outdir:结果输出目录
#我们可以通过以下函数观察都有哪些数据集可被用来富集
names = gp.get_library_name() # default: Human
names[:10]
在这里,我们选择的是KEGG_2019_Human数据集,因为NEGF是人源的细胞系
pre_res = gp.prerank(rnk=rnk, gene_sets='KEGG_2019_Mouse',
processes=4,
permutation_num=100, # reduce number to speed up testing
outdir='NEGF_gsea/prerank_report_kegg', format='png', seed=6)
输出结果被保存到了NEGF_gsea目录下
我们可以通过index查看
pre_res.res2d.sort_index()
| es | nes | pval | fdr | geneset_size | matched_size | genes | ledge_genes | |
|---|---|---|---|---|---|---|---|---|
| Term | ||||||||
| Chemokine signaling pathway | -0.767073 | -1.330912 | 0.045977 | 0.091794 | 190 | 16 | CXCL11;CCL11;CX3CL1;HCK;CXCL10;CCL20;CCL8;VAV3... | CXCL3;CXCL5;CXCL2;CXCL8;CXCL1;CXCL6 |
| Cytokine-cytokine receptor interaction | -0.702018 | -1.351795 | 0.055556 | 0.094755 | 294 | 29 | IL18;IL1RN;CXCL11;CCL11;CX3CL1;CXCL10;IL18R1;C... | LIF;CXCL3;CXCL5;CXCL2;ACKR4;IL6;IL1B;IL32;CXCL... |
| IL-17 signaling pathway | -0.801394 | -1.427337 | 0.000000 | 0.029611 | 93 | 18 | CCL11;CXCL10;CCL20;MMP9;CSF2;CSF3;CCL7;CXCL3;C... | CXCL3;CXCL5;CXCL2;IL6;IL1B;PTGS2;TNFAIP3;CXCL8... |
| NOD-like receptor signaling pathway | -0.840998 | -1.518649 | 0.000000 | 0.000000 | 178 | 15 | IL18;IRF7;NOD2;GBP4;CXCL3;CXCL2;IL6;IL1B;BIRC3... | CXCL3;CXCL2;IL6;IL1B;BIRC3;OAS1;TNFAIP3;OAS2;C... |
Term就代表了富集到的通路
2.3 单通路可视化
from gseapy.plot import gseaplot
# to save your figure, make sure that ofname is not None
gseaplot(rank_metric=pre_res.ranking, term=terms[0], **pre_res.results[terms[0]])
我们想观察每一个通路具体的富集情况,可以通过gseaplot绘制相关函数
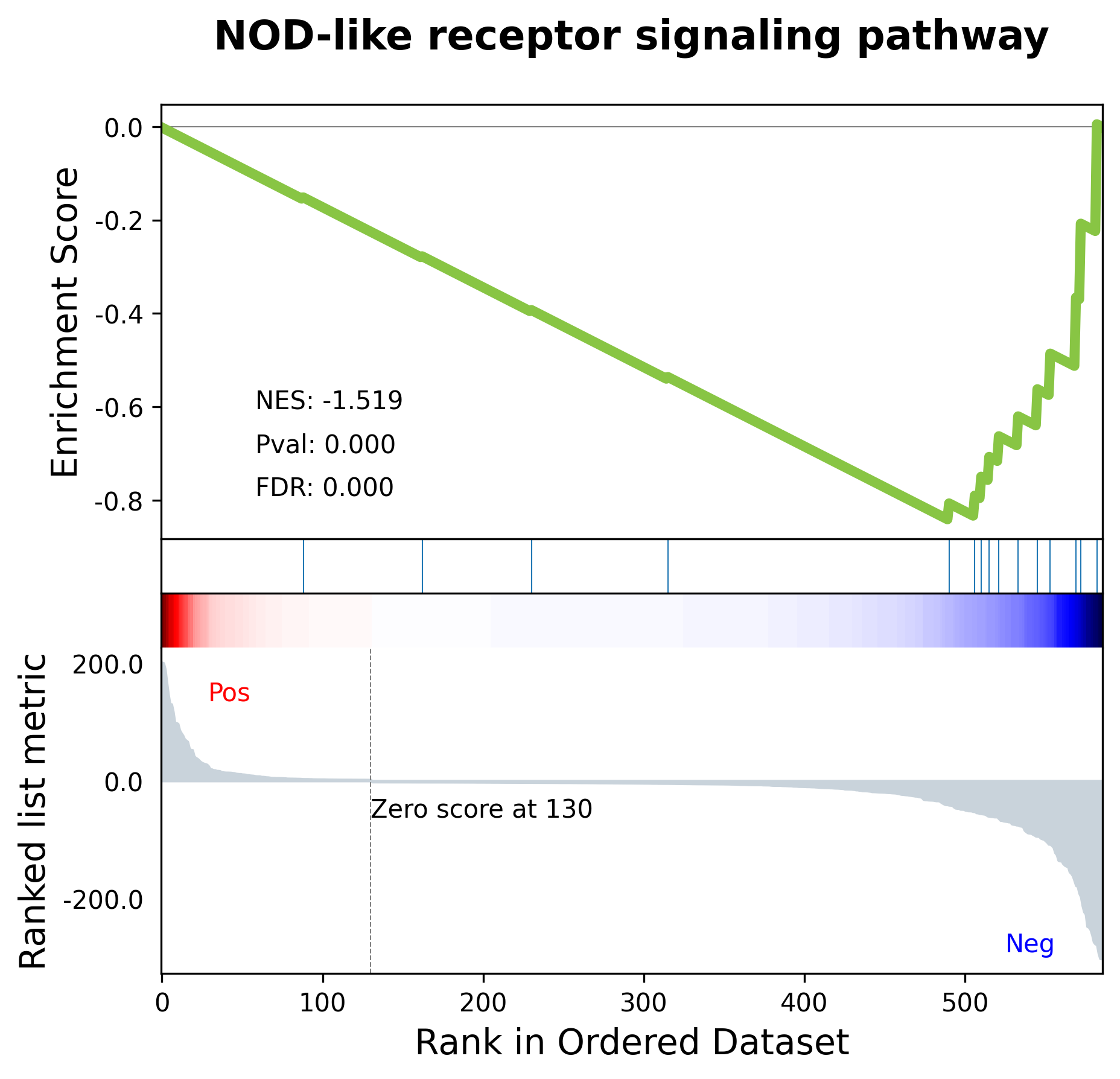
2.4 多通路可视化
我们还想观察富集到的所有通路结果,这里提供了一种气泡图的方式进行观察
sc_data=pd.read_csv(r'NEGF_gsea\prerank_report_kegg\gseapy.prerank.gene_sets.report.csv')
sc_data=sc_data.loc[sc_data['pval']<0.1]
sc_data['logp']=-np.log10(sc_data['pval']+0.001)
sc_data['com']=sc_data['matched_size']/sc_data['geneset_size']
sc_data.head()
pp=plt.figure(figsize=(3,10))
a=pp.add_subplot(1,1,1)
plt.scatter(np.zeros(len(sc_data))+1,sc_data['Term'],s=sc_data['com']*500,alpha=0.6,linewidth=2,
c=sc_data['logp'],
cmap='Greens')
plt.xticks([1],['NHDF'])
plt.xticks(range(-1,3,1))
#plt.colorbar(a)
plt.yticks(fontsize=12)
plt.xticks(fontsize=12)
plt.xlim(-0.5,1.5)
plt.savefig('NHDF_pathway.png',dpi=300,bbox_inches = 'tight')
