RNA-seq分析: 差异表达基因(DEG)分析
对于差异表达基因分析,目的是找出上面的矩阵里可能实验组跟对照组有差别的基因,有的基因在实验组中表达上调,也有可能下调,这部分基因就被我们称为差异表达基因(differential expression gene),而对于差异表达基因的计算,并不是简单的求平均值,然后比较实验组跟对照组平均值的大小,而是有一系列复杂的统计学计算在里面,但生物信息学的发展也有几十年了,研究人员已经把这些复杂的统计学封装成了函数,人们只需要调用这些函数即可。这里我们介绍的是DESeq2,一个引用突破几万的R包
1. 差异表达基因提取
1.1 数据准备
我们在上游的分析中得到了一个counts.txt,这个就是我们的基因表达矩阵,当然,你从其他任何地方得到的基因表达矩阵也可以用来进行差异表达分析~
NEGF_counts.csv
| NEGF1 | NEGF1yuan | NEGF2yuan | NEGF3 | NEGF3yuan | |
|---|---|---|---|---|---|
| Geneid | |||||
| ENSG00000223972 | 0 | 0 | 0 | 2 | 1 |
| ENSG00000227232 | 82 | 63 | 80 | 79 | 63 |
| ENSG00000278267 | 11 | 2 | 10 | 5 | 3 |
| ENSG00000243485 | 0 | 0 | 1 | 0 | 0 |
NEGF_meta.csv
我们还需要一个样本性状矩阵,来区分样本究竟是实验组还是对照组
| id | type |
|---|---|
| NEGF1 | treated |
| NEGF1yuan | control |
| NEGF2yuan | control |
| NEGF3 | treated |
| NEGF3yuan | control |
1.2 加载DESeq2
我们在R语言的环境中,输入下面一行代码
library( "DESeq2" )
1.3 导入数据
还是在R语言的环境下,我们使用read.csv函数导入table
countData <- read.csv('NEGF_counts.csv', header = TRUE, sep = ",")
metaData <- read.csv('NEGF_meta.csv', header = TRUE, sep = ",")
1.DEseq2要求输入数据是由整数组成的矩阵。
2.DESeq2要求矩阵是没有标准化的。
1.4 差异表达分析
在这一步,我们直接输入以下代码,计算差异表达矩阵,我们将结果存在dds对象中
dds <- DESeqDataSetFromMatrix(countData=countData,
colData=metaData,
design=~type, tidy = TRUE)
dds <- DESeq(dds)
表达矩阵 即上述代码中的countData,就是我们前面通过read count计算后并融合生成的矩阵,行为各个基因,列为各个样品,中间为计算reads或者fragment得到的整数。我们后面要用的是这个文件(mouse_all_count.txt)
样品信息矩阵即上述代码中的colData,它的类型是一个dataframe(数据框),第一列是样品名称,第二列是样品的处理情况(对照还是处理等),即condition,condition的类型是一个factor。这些信息可以从mouse_all_count.txt中导出,也可以自己单独建立。
差异比较矩阵 即上述代码中的design。 差异比较矩阵就是告诉差异分析函数是要从要分析哪些变量间的差异,简单说就是说明哪些是对照哪些是处理。
1.5 提取差异表达分析结果
由于dds对象并不是结果矩阵,它是DESeq2的一个数据结构,我们需要从中取出我们的差异表达分析结果,可以通过以下代码进行,结果矩阵被存放到NEGF_result1.csv
res <- results(dds)
res <- res[order(res$padj),]
write.csv(res,file='NEGF_result1.csv')
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| ENSG00000117318 | 5442.355360 | -5.710849 | 0.145082 | -39.362892 | 0.000000e+00 | 0.000000e+00 |
| ENSG00000265972 | 3147.115496 | 4.041551 | 0.103068 | 39.212444 | 0.000000e+00 | 0.000000e+00 |
| ENSG00000196611 | 4344.077177 | 4.476829 | 0.108971 | 41.082873 | 0.000000e+00 | 0.000000e+00 |
| ENSG00000125968 | 5526.691818 | -5.280627 | 0.112286 | -47.028177 | 0.000000e+00 | 0.000000e+00 |
| ENSG00000124212 | 4187.607799 | 3.441545 | 0.092530 | 37.193624 | 8.651991e-303 | 3.367009e-299 |
baseMean:(basemean_A+basemean_B)/2
log2FoldChange:差异表达倍数,实验组相对对照组而言,争执代表表达上调,负值代表表达下调
lfcSE:标准误计算
stat:统计学量
pvalue:假设检验,一般认为,p<0.05,这个基因的上下调可信
padj:对pvalue进行了一个多重假设检验
2. 基因id转换
从这一步开始,就全是Python相关的分析了,R语言在差异表达分析上的应用仅仅是计算差异表达矩阵
2.1 导入包
import pandas as pd
import matplotlib.pyplot as plt
import numpy as np
from scipy import stats
import seaborn as sns
%matplotlib inline
import mygene
mg = mygene.MyGeneInfo()
2.2 导入数据
data1=pd.read_csv('NEGF_result1.csv')
data1=data1.set_index(data1.columns[0])
data1.head()
2.3 基因id查询
在这里,我们使用了mygene这个包作为基因查询的方法,这里简单讲一下每一个参数的含义
qterms:待查询的基因list
scopes:我们查询的基因list是什么格式的,比如"entrezgene", "entrezgene,symbol",可以通过list的形式判断多种形式,如,["ensemblgene", "symbol"]。更多可在官网查询:http://mygene.info/doc/query_service.html#available_fields
fields:我们要转换的目标格式是什么格式的
species:物种名
returnall:返回查询的全部结果
data1=data1.dropna()#去重
o=mg.querymany(qterms=data1.index.values, scopes='ensembl.gene', fields='symbol', species='human',returnall=True)
输出结果
querying 1-1000...done.
querying 1001-2000...done.
querying 2001-3000...done.
……
Finished. 9 input query terms found no hit: ['ENSG00000168078', 'ENSG00000235245', 'ENSG00000287686', 'ENSG00000189144', 'ENSG00000277203', ……]
2.4 基因id转换
因为我们查询得到的list是可能会有重复,也有可能有的没查到,比如上面就有9个没查到,但我们肯定不能因为没查到就不进行基因id转换了,我们可以保留这些基因id不变,我们可以通过以下方法完成id转换的过程
2.4.1 构造待转换基因id的list
我们从查询结果中提取,并去重与忽略未查询
xl=[]#目标list
out=o['out']#查询结果
dup=[i[0] for i in o['dup']]#提取查询结果中的重复项
dup1={}#重复项存储字典,思路,如果出现了重复项就扔进字典里,第二次出现的时候就判定字典里是否出现过,出现过就认为是重复项,跳过不进行转换
for i in out:
if 'notfound' in i.keys():#未查询到,保留原始id
xl.append(i['query'])
elif 'symbol' not in i.keys():#未查询到,保留原始id
xl.append(i['query'])
elif i['query'] in dup:#查到重复项
if i['query'] in dup1.keys():#出现过就去重
continue
else:#没出现过就保留
xl.append(i['symbol'])
dup1[i['query']]=1
else:#正常转换
xl.append(i['symbol'])
len(xl)
2.4.2 对差异表达矩阵的基因id更新
可以看到,index都变成新的了
data1.index=xl
data1.head()
data1.to_csv('NEGF_data1.csv')
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| ID3 | 5442.355360 | -5.710849 | 0.145082 | -39.362892 | 0.000000e+00 | 0.000000e+00 |
| TXNIP | 3147.115496 | 4.041551 | 0.103068 | 39.212444 | 0.000000e+00 | 0.000000e+00 |
| MMP1 | 4344.077177 | 4.476829 | 0.108971 | 41.082873 | 0.000000e+00 | 0.000000e+00 |
| ID1 | 5526.691818 | -5.280627 | 0.112286 | -47.028177 | 0.000000e+00 | 0.000000e+00 |
| PTGIS | 4187.607799 | 3.441545 | 0.092530 | 37.193624 | 8.651991e-303 | 3.367009e-299 |
3. 差异基因可视化
我们在前面的分析中得到了差异表达基因的计算结果,但是现在出现一个新的问题,那就是具体哪些基因才是我们感兴趣的差异表达基因?我们该用什么方法确定呢?
首先,我们观察前面的res发现log2FoldChange这一列,这一列就是代表实验组相对对照组而言,基因表达水平的倍数变化,一般来说,我们会通过直方图的方式,来确定合适的表达倍数阈值。
3.1 差异表达倍数选取
我们通过一行简单的代码,观察表达倍数的分布
#绘制直方图
plt.hist(data1['log2FoldChange'])
输出结果如图,从图中我们发现,2或许是一个不错的阈值,也就是说,当实验组基因的表达是对照组的两倍的时候,我们认为这是表达上调的,下调同理。
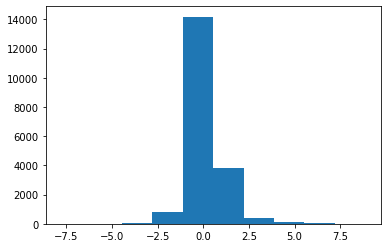
3.2 差异表达基因标记
在前面的分析中,我们确定了log2FoldChange的阈值为2,那么我们就将表达变化大于2的基因全部标记上up跟down,注意,这里还有一个标准,就是padj<0.05。
data1['sig'] = 'normal'
data1.loc[(data1.log2FoldChange> 2 )&(data1.padj < 0.05),'sig'] = 'up'
data1.loc[(data1.log2FoldChange< -2 )&(data1.padj < 0.05),'sig'] = 'down'
data1['log(padj)'] = -np.log10(data1['padj'])
data1.head()
观察数据的输出,我们发现,所有的基因都被标记上了类别
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | sig | log(padj) | |
|---|---|---|---|---|---|---|---|---|
| ID3 | 5442.355360 | -5.710849 | 0.145082 | -39.362892 | 0.000000e+00 | 0.000000e+00 | down | inf |
| TXNIP | 3147.115496 | 4.041551 | 0.103068 | 39.212444 | 0.000000e+00 | 0.000000e+00 | up | inf |
| MMP1 | 4344.077177 | 4.476829 | 0.108971 | 41.082873 | 0.000000e+00 | 0.000000e+00 | up | inf |
| ID1 | 5526.691818 | -5.280627 | 0.112286 | -47.028177 | 0.000000e+00 | 0.000000e+00 | down | inf |
| PTGIS | 4187.607799 | 3.441545 | 0.092530 | 37.193624 | 8.651991e-303 | 3.367009e-299 | up | 298.472756 |
3.3 差异表达基因可视化
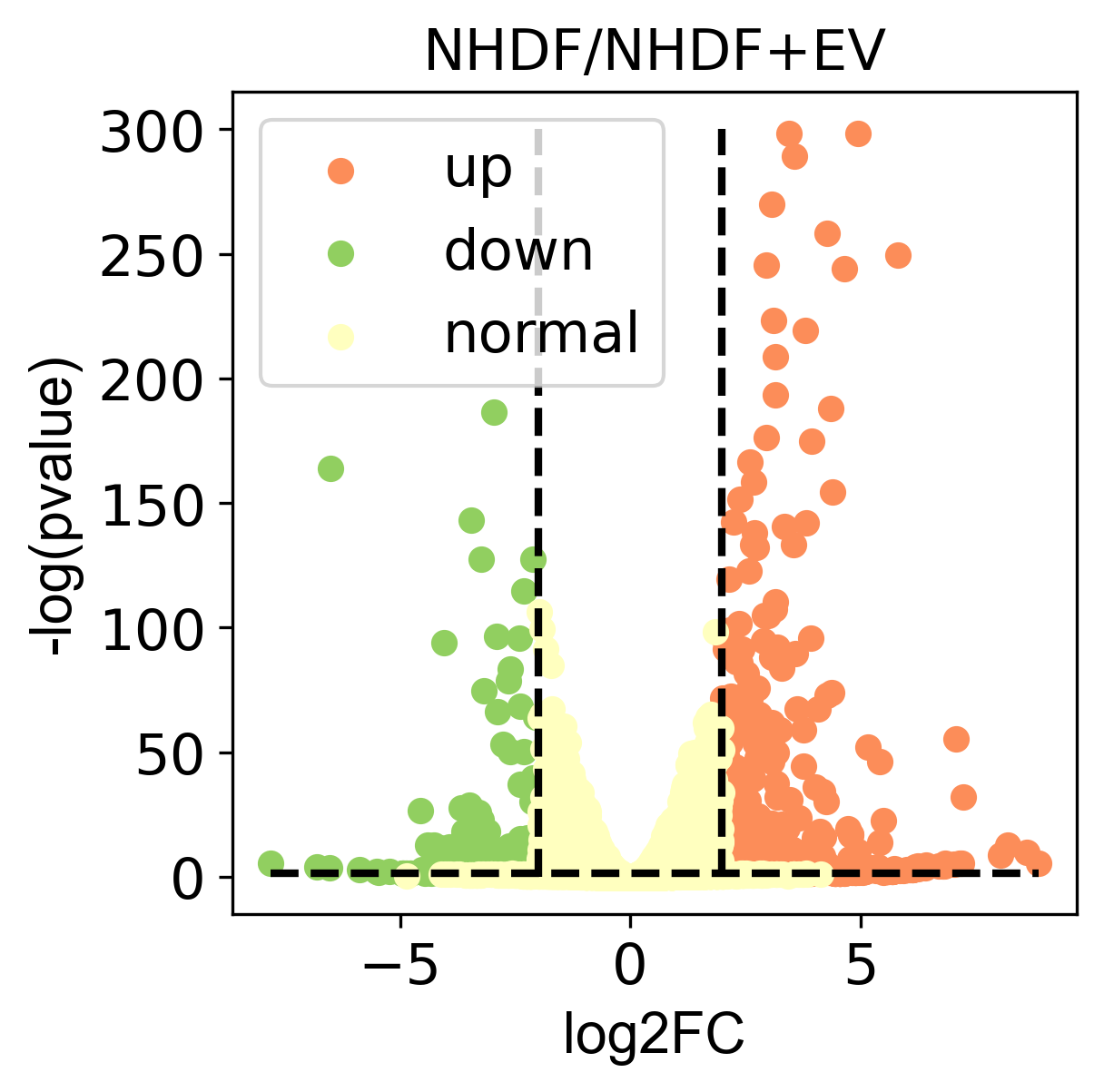
我们现在已经把差异表达的基因标记完成了,下一步,就是看看差异表达基因的分布了。在这里,我们会用一种叫火山图的方式展示数据,方法如下
#定义字体
font1={
'font':'Arial',
'size':15,
}
#定义图片大小
pp=plt.figure(figsize=(4,4))
#用ax控制图片
ax=pp.add_subplot(1,1,1)
#绘制表达上调的基因
plt.scatter(x=data1[data1['sig']=='up']['log2FoldChange'],y=data1[data1['sig']=='up']['log(padj)'],color='#FC8D59',label='up')
#绘制表达下调的基因
plt.scatter(x=data1[data1['sig']=='down']['log2FoldChange'],y=data1[data1['sig']=='down']['log(padj)'],color='#91CF60',label='down')
#绘制非差异表达基因
plt.scatter(x=data1[data1['sig']=='normal']['log2FoldChange'],y=data1[data1['sig']=='normal']['log(padj)'],color='#FFFFBF',label='normal')
#设置横纵坐标轴字体
plt.yticks(fontsize=15)
plt.xticks(fontsize=15)
#设置图注
plt.legend(loc='best',fontsize=15)
#设置横纵标题
ax.set_ylabel('-log(pvalue)',font1)
ax.set_xlabel('log2FC',font1)
#绘制虚线,padj
ax.plot([data1['log2FoldChange'].min(),data1['log2FoldChange'].max()],[-np.log10(0.05),-np.log10(0.05)],linewidth=2, linestyle="--",color='black')
#绘制虚线,表达上调分割线
ax.plot([2,2],[data1['log(padj)'].min(),300],linewidth=2, linestyle="--",color='black')
#绘制虚线,表达下调分割线
ax.plot([-2,-2],[data1['log(padj)'].min(),300],linewidth=2, linestyle="--",color='black')
#绘制图像标题
plt.title('NHDF/NHDF+EV',fontsize=15)
#保存图片
plt.savefig("NHDF_volcano.png",dpi=300,bbox_inches = 'tight')
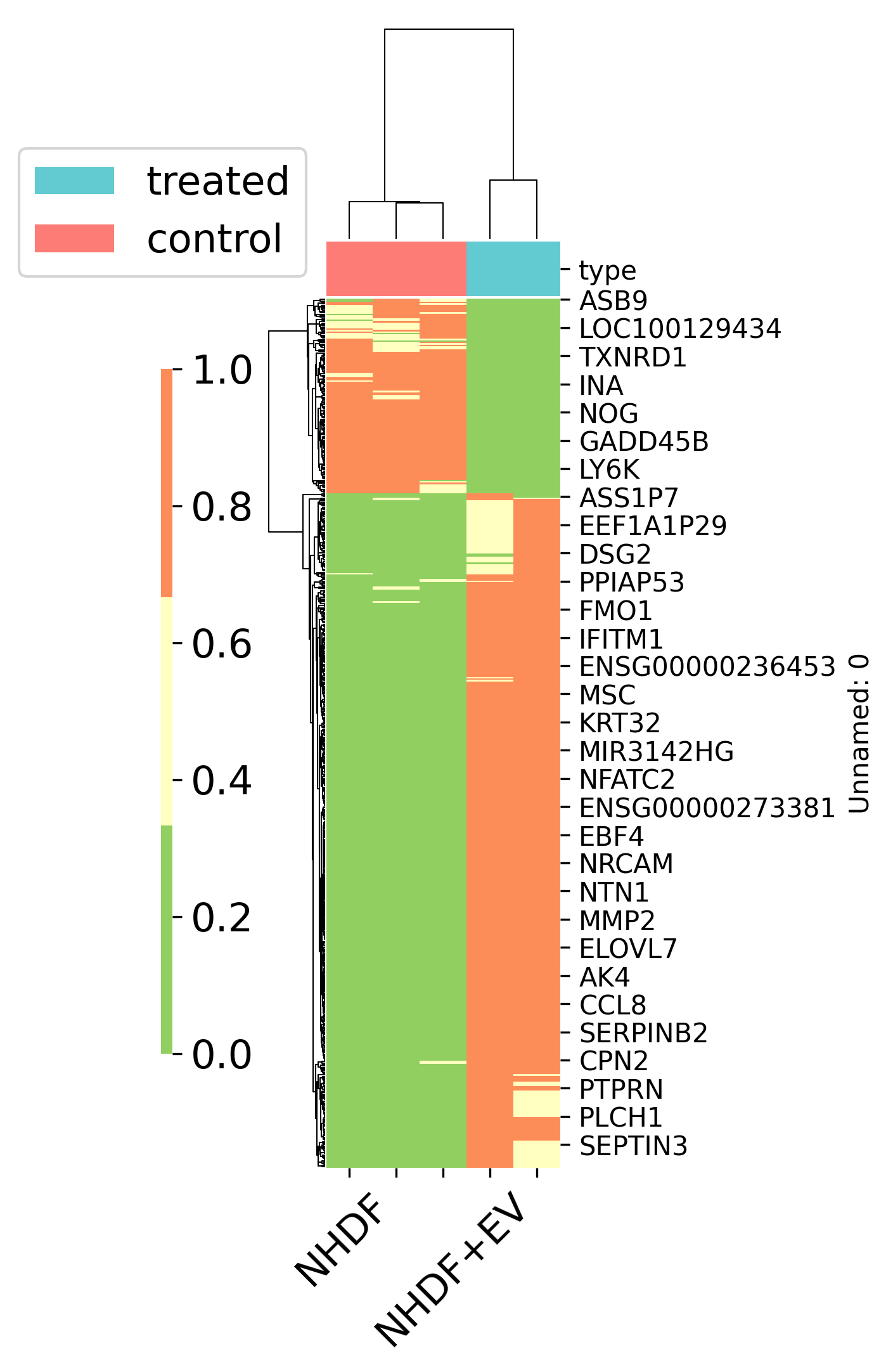
4. 差异表达热图
我们现在得到了差异表达基因的分布,那么我们还得进一步确定的是,这些差异表达的基因在每个样本中,是否是真的表达上调或表达下调呢,我们这里会借助热图的方式来对差异表达基因进行可视化。
4.1 导入数据
#导入差异分析原始结果(未转换基因id)
data2=pd.read_csv('NEGF_result1.csv')
data2=data2.set_index(data2.columns[0])
#导入原始counts矩阵
data=pd.read_csv('NEGF_counts.csv')
data.set_index(data.columns[0],inplace=True)
4.2 提取差异表达基因的原始counts矩阵
filtered=data.loc[data2.dropna().index]
filtered.index=xl
filtered.head()
结果如下
| NEGF1 | NEGF1yuan | NEGF2yuan | NEGF3 | NEGF3yuan | |
|---|---|---|---|---|---|
| ID3 | 181 | 8455 | 10398 | 140 | 9382 |
| TXNIP | 6719 | 445 | 427 | 6833 | 513 |
| MMP1 | 9307 | 551 | 426 | 9820 | 468 |
| ID1 | 207 | 9829 | 8904 | 231 | 9901 |
| PTGIS | 8603 | 897 | 838 | 8684 | 942 |
提取差异表达基因
res=data2.dropna()
res.index=xl
res['log(padj)'] = -np.log10(res['padj'])
res['sig'] = 'normal'
res.loc[(res.log2FoldChange> 2 )&(res.padj < 0.05),'sig'] = 'up'
res.loc[(res.log2FoldChange< -2 )&(res.padj < 0.05),'sig'] = 'down'
filtered_ids=res.loc[res['sig']!='normal'].index
len(filtered.loc[filtered_ids])
filtered.loc[filtered_ids].to_csv('NEGF_heatmap_data.csv')
4.3 可视化
我们得到差异表达基因的原始表达矩阵后,就可以开始对这些基因进行可视化了
4.3.1 数据准备
我们需要准备一个meta矩阵跟hetamap_data矩阵,这两个矩阵在前面的分析已经准备好了。
4.3.2 导入数据
NEGF_heatmap_data=pd.read_csv('NEGF_heatmap_data.csv')
NEGF_heatmap_data.set_index(NEGF_heatmap_data.columns[0],inplace=True)
4.3.3 热图
import palettable
#导入数据
meta=pd.read_csv('NEGF_meta.csv')
meta=meta.set_index(meta.columns[0])
#设置组别对应的颜色
col_c={
'control':'#FD7C76',
'treated':'#62CAD1'
}
#绘制聚类热图
a=sns.clustermap(NEGF_heatmap_data,
cmap=palettable.colorbrewer.diverging.RdYlGn_3_r.mpl_colors,
standard_scale = 0,figsize=(2,8),
col_colors=meta['type'].map(col_c),
)
#设置聚类热图的横纵坐标标签大小
a.ax_heatmap.yaxis.set_tick_params(labelsize=10)
a.ax_heatmap.xaxis.set_tick_params(labelsize=15)
#设置聚类热图横坐标的内容
a.ax_heatmap.xaxis.set_ticklabels(['','NHDF','','','NHDF+EV'])
#设置聚类热图横坐标的旋转角度
labels=a.ax_heatmap.xaxis.get_ticklabels()
#a.ax_heatmap.xaxis.set_text(['','RAW','','','RAW+EV',''])
plt.setp(labels, rotation=45, horizontalalignment='right',)
#绘制组别颜色条
for label in meta['type'].unique():
a.ax_col_dendrogram.bar(0, 0, color=col_c[label],
label=label, linewidth=0)
#绘制组别颜色条的legend
a.ax_col_dendrogram.legend(loc="best", ncol=1,bbox_to_anchor=(-0.5, 0., 0.5, 0.5),fontsize=15)
#设置legend的位置
a.cax.set_position([-.15, .2, .03, .45])
#设置颜色条的坐标大小
plt.setp(a.cax.yaxis.get_majorticklabels(), fontsize=15)
#保存图片
plt.savefig("NHDF_heatmap.png",dpi=300,bbox_inches = 'tight')
结果如图所示